
Production and Evaluation of Lipid Nanoparticle as Drug Carrier for
Treatment of Alzheimer’s Disease to Enhance the Blood Brain
Barrier Penetration
S. Chitra, A. Sundar Raj, G. Yalini and K. Raghuram
Department of Biomedical Engineering, E.G.S Pillay Engineering College, Nagapattinam - 611002, Tamil Nadu, India
Keywords: Alzheimer's Disease, Lipid Nanoparticles, Blood‑Brain Barrier, Drug Delivery, Amyloid‑Beta, Tau Protein,
Neuroinflammation, Therapeutic Applications, siRNA, Sustained Release.
Abstract: Alzheimer's disease (AD) is a progressive neurodegenerative disorder that most frequently affects older
adults, resulting in cognitive impairment, memory loss, and behavioural changes. It is the most prevalent
cause of dementia and an increasing global health issue with aging populations. The pathophysiology of AD
is marked by amyloid-beta plaque deposition, tau protein tangle formation, and synaptic loss, leading to
neuronal dysfunction and cell death. Despite the fact that existing pharmacological therapies may correct
symptoms, they do not reverse or stop disease progression. One of the greatest obstacles to treating AD is the
blood-brain barrier (BBB), which shields the brain from the effective delivery of therapeutic compounds and
thus reduces the effectiveness of most drugs. This restriction necessitates novel drug delivery systems that
have the ability to cross the BBB and deliver drug therapeutic agents in adequate concentrations into the brain.
Lipid nanoparticles (LNPs) also present a potent solution to such a challenge. LNPs represent nanosized lipid-
based carrier systems that encapsulate hydrophobic and hydrophilic drugs, providing an environment of a
biocompatible platform for delivery of drugs as targeted therapy. The inherent characteristics of LNPs,
including their nanoscale dimensions, flexibility, and capacity to alter surface chemistry, make them capable
of interacting favourably with biological membranes and penetrating the BBB. This is a quality that makes
LNPs especially ideal for delivering therapeutic agents to the brain. The therapeutic uses of LNPs in the
treatment of AD are multifaceted, targeting the most important pathological characteristics of the disease,
including amyloid-beta plaques and tau protein tangles. LNPs may be used to deliver small molecules,
peptides, and antibodies to prevent amyloid-beta aggregation or facilitate clearance of amyloid-beta from the
brain, perhaps delaying disease. RNA therapies such as siRNA and antisense oligonucleotides are deliverable
through LNPs and can suppress production of amyloid-beta, serving as an alternate mechanism for
modification of disease. Overall, lipid nanoparticles are a promising drug delivery system for Alzheimer's
disease. Their capacity to penetrate the BBB, reach specific areas of the brain, and deliver therapeutic agents
in controlled release makes them an important resource for treating the intricacies of AD.
1 INTRODUCTION
1.1 Overview of Alzheimer's Disease
Alzheimer's disease (AD) is a chronic, progressive
neurodegenerative disease that causes cognitive
impairment, such as memory loss, compromised
reasoning, and problem-solving difficulty. It is the
most common cause of dementia, a syndrome of
profound loss of cognitive function, which has a
significant impact on an individual's capability to
perform activities of daily living. Alzheimer's disease
most commonly affects older individuals, and its
incidence rises with an aging population across the
globe. Estimates suggest that by the year 2050,
Alzheimer's disease will double the number of
individuals with the disease, and thus it will be an
enormous strain on the public health care systems
globally (Cummings et al., 2019). The disease is
accompanied by initial mild forgetfulness in its early
stage, but subsequently, the patient develops critical
memory loss, language problems, and confusion. In
its advanced stages, the patient loses the ability to
recognize close relatives, becomes bedridden, and
needs 24-hour care (Cunningham, C et al., 2020). At
168
Chitra, S., Raj, A. S., Yalini, G. and Raghuram, K.
Production and Evaluation of Lipid Nanoparticle as Drug Carrier for Treatment of Alzheimer’s Disease to Enhance the Blood Brain Barrier Penetration.
DOI: 10.5220/0013924500004919
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 1st International Conference on Research and Development in Information, Communication, and Computing Technologies (ICRDICCT‘25 2025) - Volume 5, pages
168-181
ISBN: 978-989-758-777-1
Proceedings Copyright © 2026 by SCITEPRESS – Science and Technology Publications, Lda.

the molecular level, AD is defined by the presence of
amyloid-beta (Aβ) plaques deposits, tau protein
tangles, and neuroinflammation, which all lead to
neuronal dysfunction and cell death. The amyloid
plaques, composed of the aggregation of Aβ peptides,
block synaptic communication by disrupting
neuronal signalling (Cheng, S et al., 2020).
Meanwhile, tau tangles, due to the
hyperphosphorylation of tau proteins, disrupt
microtubule stability and intracellular transport,
further destroying neurons and inducing cognitive
impairment (Yuan et al., 2020). Despite years of
investigation into possible treatments, AD therapies
currently remain primarily symptomatic.
Acetylcholinesterase inhibitors like donepezil and
rivastigmine, which increase the concentration of
acetylcholine in the brain, have modest benefits in
improving performance on memory tasks and
reducing cognitive decline (Zhou et al., 2018).
Similarly, glutamate receptor antagonists like
memantine aim to block excitotoxicity through the
regulation of glutamate transmission, but these drugs
fail to treat the root causes of the disease (Cummings
et al., 2019). Thus, disease-modifying treatments that
not only alleviate symptoms but also reverse or slow
down AD progression are in dire need. A major
roadblock to identifying effective therapies is the
blood-brain barrier (BBB), a physical barrier that
impedes the delivery of therapeutic compounds into
the brain.
1.2 Blood-Brain Barrier (BBB) Issue
with Alzheimer's Therapy
The blood-brain barrier (BBB) is a selectively
permeable membrane between the circulatory system
and the brain. It is formed by tight junctions of
endothelial cells, pericytes, and astrocyte end-feet,
which collectively exclude potentially toxic
substances while allowing the entry of necessary
nutrients and gases (Zhou et al., 2018). The BBB is
an essential protective system for the brain, protecting
it from toxins, infection, and alterations in blood
composition. But the same factors that render the
BBB such a powerful protective system render very
significant obstacles to the delivery of drugs to the
brain. While the BBB allows small molecules such as
glucose and oxygen to pass across, it actively
excludes the passage of large molecules, including
most therapeutic drugs that might potentially be of
value in the treatment of neurological disorders such
as Alzheimer's (Zhou et al., 2018). In Alzheimer's, the
BBB poses another barrier: many drug candidates,
even amyloid-beta plaque, tau tangles, and
neuroinflammation candidates, are unable to cross the
BBB in therapeutic levels (Wang et al., 2022). As a
result, even potential candidates cannot reach their
target site in the brain, greatly hindering the
development of effective treatments. This is
compounded by the fact that AD pathophysiology
involves more than one molecular mechanism, such
as amyloid-beta deposition, tau
hyperphosphorylation, oxidative stress, and
neuroinflammation, all of which require targeted drug
delivery systems that can access specific regions of
the brain (Zhang et al., 2019). BBB penetration has
been a prominent area of study in drug delivery for
Alzheimer's disease. Different strategies have been
proposed, including the use of focused ultrasound for
reversible disruption of the BBB, receptor-mediated
delivery, and nanoparticle-based drug delivery
systems (Li et al., 2021). While some of these
strategies have seemed promising, they are typically
marred by problems of invasiveness, efficacy, and
safety. Of these strategies, lipid nanoparticles (LNPs)
have garnered a lot of attention as a promising
strategy to drug delivery to the brain. The unique
features of LNPs, including their nanoscale size,
biocompatibility, and capacity to encapsulate a range
of therapeutic agents, make them most suited to cross
the BBB and deliver drugs directly to the brain (Song
et al., 2020).
1.3 Use of Lipid Nanoparticles (LNPs)
in Drug Delivery
Lipid nanoparticles (LNPs) are nanocarriers that have
been extensively studied for their ability to deliver
therapeutic agents through the BBB and into the
brain. LNPs are typically composed of lipid
molecules in a formulation that is able to encapsulate
hydrophobic as well as hydrophilic drugs, thus
making them universal carriers of a range of
therapeutic compounds (Li et al., 2021). Lipid
composition of LNPs provides some benefits such as
biocompatibility, biodegradability, and reduced
toxicity, all of which play crucial roles in achieving
the successful delivery of drugs into the brain (Kalluri
et al., 2019). The strongest advantage of LNPs is also
their size, typically ranging between 20-100 nm. This
size helps LNPs move across the junctions of BBB
endothelial cells by mechanisms of endocytosis or
transcytosis (Li et al., 2021). The figure 1 shows the
Pictorial Representation of Lipid Nanoparticle (LNP)
for Alzheimer's Disease Treatment (Song et al.,
2020). The ability of LNPs to traverse the BBB and
deliver medicines to the brain in a specific manner is
useful particularly in diseases of the nerve system
Production and Evaluation of Lipid Nanoparticle as Drug Carrier for Treatment of Alzheimer’s Disease to Enhance the Blood Brain Barrier
Penetration
169

such as Alzheimer's disease, where selective delivery
is pivotal in order to achieve therapeutic performance.
Moreover, LNPs are engineered to provide a broad
selection of therapeutic species such as peptides,
proteins, nucleic acids, and small molecules of
applicability to therapy of various aspects of
Alzheimer's pathophysiology (Wang et al., 2022). In
addition to the ability to provide a variety of
therapeutic drugs, LNPs also have controlled and
sustained release. This is particularly critical in the
treatment of long-term conditions like Alzheimer's,
where stable drug levels in the brain over extended
timeframes can optimize therapeutic effects and
reduce dosing frequency (Song et al., 2020). With
controlled release of drug-encapsulated drugs, LNPs
can ensure that the drug is active in the brain for
extended durations without therapeutic failure caused
by ineffective drug levels.
Figure 1: Pictorial representation of lipid nanoparticle
(LNP) for Alzheimer's disease treatment (Song et al., 2020).
1.4 Therapeutic Applications of Lipid
Nanoparticles for the Treatment of
Alzheimer's Disease
Lipid nanoparticles (LNPs) have emerged to be
extremely promising in the treatment of several
therapeutic problems in Alzheimer's disease.
Amyloid-beta (Aβ), a peptide that aggregates to
deposit as plaques in the brain, disrupting synaptic
function and causing cognitive impairment, is one of
the principal therapeutic targets in AD. Aβ plaques
are one of the first and most spectacular
manifestations of AD pathology and are therefore a
significant target for drug treatments. Recent studies
have established the ability of LNPs to deliver
therapeutic cargoes, such as small molecule
inhibitors, antibodies, or RNA-based therapies, which
could prevent amyloid-beta aggregation or enable its
clearance from the brain (Yuan et al., 2020). These
approaches are intended to prevent amyloid-beta
toxicity on neurons and restoration of normal synaptic
function. Besides modulation of amyloid-beta
plaques, tau protein is another prime target of AD. Tau
form neurofibrillary tangles in neurons, leading to
intracellular transport disruption and
neurodegeneration. Tau tangles are also involved in
later stages of the disease and are believed to be at the
centre of cognitive impairment (Zhang et al., 2020).
LNPs can be used to deliver small molecule inhibitors
or RNA-based medicine that modulates tau
phosphorylation, aggregation, and clearance.
Through tau targeting, the researchers hope to
suppress or even reverse tau-mediated
neurodegeneration, offering a putative disease-
modifying treatment for Alzheimer's patients (Li et
al., 2021). They also suggest that neuroinflammation
is a key contributor to Alzheimer's pathogenesis.
Persistent brain inflammation exacerbates neuronal
damage and accelerates disease progression.
Activated microglia and inflammatory cytokines are
typically elevated in AD patient brains, which is
implicated in the neurodegenerative process (Kalluri
et al., 2020). LNPs can be utilized to deliver anti-
inflammatory medication to the brain, reducing
neuroinflammation and its harmful effects. This can
potentially slow disease progression and improve
cognitive function in Alzheimer's patients. The
flexibility of LNPs makes them amenable to deliver a
wide variety of therapeutic agents that can target
various aspects of Alzheimer's disease pathology.
Besides amyloid-beta, tau, and neuroinflammation,
LNPs can be designed to deliver drugs that modulate
neurotransmitter levels, enhance synaptic plasticity,
or provide neuroprotection against oxidative stress.
These multi-target approaches are likely to offer the
most effective way to treat Alzheimer's disease since
they can effectively address the multifactorial and
complex nature of the disease (Song et al., 2020).
2 LITERATURE REVIEW
2.1 Pathophysiology of Alzheimer's
Disease and Therapeutic
Challenges
Alzheimer's disease (AD) is a complex multifactorial
neurodegenerative disorder causing progressive
cognitive impairment, ultimately disabling the
individual's ability to perform activities of daily
living. The most prevalent form of dementia,
Alzheimer's disease affects millions of individuals
globally, and projections are that the number of cases
will swell exponentially within the next few decades
as the global population ages. AD typically begins
with the insidious and gradual onset of memory
impairment, accompanied by language deficits,
ICRDICCT‘25 2025 - INTERNATIONAL CONFERENCE ON RESEARCH AND DEVELOPMENT IN INFORMATION,
COMMUNICATION, AND COMPUTING TECHNOLOGIES
170

spatial disorientation, and deficits in executive
function. The clinical course is one of slow
deterioration of cognitive function, with the earliest
and most prominent of symptoms being loss of
memory. The disease also impairs other cognitive
areas, including reasoning, problem-solving, and
decision-making.
At the pathological level, the two characteristic
features of AD are extracellular amyloid-beta (Aβ)
plaques and intracellular tau neurofibrillary tangles,
both of which disrupt normal neuronal function and
are responsible for the neurodegenerative process.
Amyloid plaques are composed of aggregates of
amyloid-beta peptide, which is the product of
aberrant cleavage of amyloid precursor protein (APP)
by enzymes like beta-secretase and gamma-secretase.
Such a build-up of plaques disrupts synaptic
transmission, which disrupts neural circuits, most
significantly in areas like the hippocampus and
cortex, which are essential to memory and cognition
functions (Cheng, S et al., 2020), (Yuan et al., 2020).
Alternatively, tau tangles resulting from tau protein
hyperphosphorylation also lead to neuronal
dysfunction. Tau is a microtubule-associated protein
that stabilizes the microtubule structure of the neuron
and promotes organelle and nutrient transport. In AD,
however, tau is abnormally hyperphosphorylated and,
in doing so, loses its microtubule association and
instead forms aggregates in the neuron to create
twisted tangles. These tangles disrupt neuronal
transport and lead to the cell death of affected
neurons, ultimately resulting in brain atrophy and the
resulting cognitive impairments (Yuan et al., 2020),
(Zhou et al., 2018).
Besides amyloid plaques and tau tangles,
neuroinflammation is another key mechanism in the
pathogenesis of AD. Neuroinflammation is the
activation of glial cells such as astrocytes and
microglia following neuronal damage. While glial
cells play a key role in ensuring homeostasis in the
brain, chronic glial cell activation is the reason behind
the release of pro-inflammatory cytokines and
reactive oxygen species, which also harm neurons
and promote neurodegeneration. Recent findings
have implicated the possibility of targeting
neuroinflammation as a potential way of reducing the
impact of AD and slowing the progression of the
disease (Zhou et al., 2018), (Wang et al., 2022).
While pathological mechanisms of AD are well
characterized, therapeutic interventions are
symptomatic. Currently approved drugs, e.g.,
acetylcholinesterase inhibitors (donepezil,
rivastigmine, and galantamine), act by increasing the
concentration of acetylcholine in the brain, a
neurotransmitter involved in learning and memory.
These drugs are not disease etiology curative but at
best modestly effective in slowing the rate of
cognitive decline. A second class of drugs, glutamate
modulators like memantine, decreases excitotoxicity
by modulating glutamate neurotransmission but, like
the first, is symptomatic only and does not alter the
course of the disease (Wang et al., 2022). The lack of
useful disease-modifying therapies is due to the
multifactorial and complicated etiology of AD, not
caused by a single but by the synergistic pathogenic
interaction of genetic, environmental, and lifestyle
factors. The reality of current drug discovery is
plagued with challenges in the ability to discover
molecular targets that can retard or halt disease
progression, and in the ability to provide assurance
that potential therapeutic agents can enter the brain.
The blood-brain barrier (BBB), a selective membrane
to protect the brain from toxic substances, is prone to
bar the effective delivery of drugs and biological
mediators, such as proteins, antibodies, and small
molecules. Therefore, the development of novel
therapeutic strategies to AD requires novel drug
delivery systems with the capability to traverse this
barrier (Zhang et al., 2019), (Song et al., 2020).
2.2 Drug Delivery and Blood-Brain
Barrier Issues
The blood-brain barrier (BBB) is a selective
semipermeable membrane that protects the CNS from
toxins and pathogens but does allow necessary
nutrients to pass through. While it serves a protective
role, however, the BBB is a significant barrier to the
delivery of drugs to the brain. BBB is made up of
pericytes, endothelial cells, and astrocytic end-feet,
which have tight junctions among them that limit the
diffusion of charged entities and large molecules from
the blood into the brain. Thus, many promising drugs
for the treatment of Alzheimer's disease and other
neurodegenerative disorders find it difficult to cross
the BBB in sufficient quantities to be effective (Song
et al., 2020), (Li et al., 2021).
Several strategies have been suggested to enhance
drug delivery through the BBB. One is to create drugs
that are sufficiently small or lipophilic to pass through
the BBB by passive diffusion. But this is typically not
sufficient, as many therapeutic compounds, like
antibodies, small molecules, and nucleic acids, are
too large or hydrophilic to pass through the BBB on
their own. A second strategy is to temporarily open up
the BBB via methods like focused ultrasound or
osmotic disruption so that drugs can travel more
Production and Evaluation of Lipid Nanoparticle as Drug Carrier for Treatment of Alzheimer’s Disease to Enhance the Blood Brain Barrier
Penetration
171

easily into the brain. But these methods are invasive,
and long-term safety is unknown (Li et al., 2021).
One of the most promising alternative approaches is
the application of drug delivery systems, such as
nanoparticles, liposomes, and viral vectors, in an
attempt to be designed to cross the BBB and deliver
therapeutic substances to the brain. Of these, lipid
nanoparticles (LNPs) have been of interest because
they have the potential to circumvent the BBB and
deliver a broad range of therapeutic molecules, such
as small molecules, proteins, and nucleic acids. The
small particle size of LNPs (range 20-100 nm)
ensures that they are able to traverse the BBB by
passive diffusion or receptor-mediated endocytosis.
LNPs can even be targeted by using targeting ligands
that direct them to areas in the brain in AD pathology
(Zhang et al., 2020), (Kalluri et al., 2019).
Lipid nanoparticles are also very promising because
they are biocompatible and biodegradable and thus
are ideally suited for long-term application in the
treatment of neurodegenerative disorders. The lipid
part of LNPs is typically derived from naturally
occurring lipids, such that the particles become safe
for use in humans. Further, the fact that LNPs can
entrap hydrophilic and hydrophobic drugs makes
them a simple drug delivery vehicle. They can be
employed to deliver a range of therapeutic drugs such
as small molecules, RNA therapeutics, and proteins to
the brain and provide an alternative non-invasive and
effective route of treatment (Yuan et al., 2020),
(Alzheimer et al. 2020).
2.3 Lipid Nanoparticles: Design and
Properties
Lipid nanoparticles (LNPs) are nanoscale drug
delivery systems composed primarily of lipid
components, used to encapsulate drugs and deliver
them to target tissues or organs. An LNP typically has
a lipid core to wrap around the drug load, and a
surfactant or excipient shell to stabilize the particle.
This composition allows LNPs to wrap a broad range
of therapeutic agents, from hydrophobic molecules to
small molecules, nucleic acids, and proteins.
Among the significant advantages of LNPs is that
they can cross the blood-brain barrier. Particle size in
the case of LNPs constitutes one of the most critical
parameters for BBB crossing. The nanoparticles of
the size range of 20-100 nm are more effective in
crossing the BBB because they possess the ability to
bind to endothelial cells and get internalized by
receptor-mediated endocytosis. Surface charge of the
LNPs is an important determinant for BBB crossing.
Cationic nanoparticles will be more likely to
penetrate the BBB at a higher rate due to electrostatic
attraction with negatively charged endothelial cells
lining the brain's blood vessels (Kalluri et al., 2019),
(Yuan et al., 2020).
Beyond their ability to traverse the BBB, LNPs may
be designed to produce therapeutic action at specific
areas of the brain that are impacted by Alzheimer's
disease. This can be achieved through surface
functionalization of the nanoparticles with targeting
ligands, which bind to the receptors that are
overexpressed in brain areas impacted by amyloid-
beta plaques or tau tangles. Targeting ligands such as
antibodies, peptides, or aptamers may be surface
functionalized onto the LNPs to make them more
targeted and selective towards specific brain areas
(Yuan et al., 2020), (Alzheimer et al. 2020). Besides
that, LNPs are highly biocompatible and
biodegradable, and hence can be employed in drug
delivery for an extended period of time without
inflicting any harm. The lipid components used in
LNPs are natural, and hence the risk of toxicity or
immunogenicity is minimized. Furthermore, the lipid
structure of LNPs is easily modifiable to improve
their drug delivery properties, such as stability,
release kinetics, and targeting capacity (Alzheimer et
al. 2020) (Wang, et al. 2020).
2.4 In Vivo Investigations of Lipid
Nanoparticles for Alzheimer's
Disease
Several preclinical studies have shown the
therapeutic potential of lipid nanoparticles in
delivering therapeutic agents to the brain and
improving cognitive function in animal models of
Alzheimer's disease. LNPs have been used to deliver
various therapeutic agents, including small molecule
inhibitors, antibodies, and RNA therapeutics, to the
brain. A study showed that LNPs could deliver anti-
amyloid-beta antibodies effectively to the animal
models, decreasing amyloid plaque burden and
memory performance. These results suggest that
LNPs can be used as an efficient drug delivery system
for amyloid-targeted therapy in AD (Zhang et al.,
2020), (Li et al., 2021). Apart from small molecules
and antibodies, LNPs have been used for delivery of
RNA therapeutics, such as siRNA and mRNA, in
Alzheimer's disease models. These RNA drugs can be
designed to selectively target genes that encode
amyloid-beta or tau to correct for the disease-causing
factors. For example, LNPs encapsulating siRNA for
tau have been effective in reducing tau pathology and
preventing neurodegeneration in preclinical AD
models. This treatment represents a novel
ICRDICCT‘25 2025 - INTERNATIONAL CONFERENCE ON RESEARCH AND DEVELOPMENT IN INFORMATION,
COMMUNICATION, AND COMPUTING TECHNOLOGIES
172

intervention in the treatment of AD through
regulation of the molecular pathways of disease
pathology (Zhang et al., 2020), (Kalluri et al., 2020).
Another possible application of LNPs for AD is in
gene therapy. By introducing genetic material, such
as genes encoding therapeutic proteins, directly into
the brain, LNPs could potentially reverse the course
of AD and restore normal brain function. For
instance, LNPs have been used to deliver genes
encoding anti-inflammatory cytokines or
neurotrophic factors to promote neuronal survival and
suppress neuroinflammation. Such approaches also
have great promise for the development of disease-
modifying drugs that both offer symptomatic relief
and affect the underlying causes of AD pathology
(Zhang, et al. 2021).
3 MATERIALS AND
SPECIFICATIONS
3.1 High-Shear Homogenizer
High-shear homogenizer is a critical instrument in the
production of lipid nanoparticles (LNPs). It is
employed mainly for emulsification and dispersion of
the lipid-therapeutic agent blend to obtain uniform
particle size and composition. The homogenizer
functions by subjecting the lipid-API blend to
mechanical shear forces, which disperse it into
nanoscale droplets. The equipment is usually run at a
pressure of 500–2000 bar and a flow rate of 10–50
mL/min, depending on the production scale. The
high-speed rotor or impeller creates shear forces, and
the regulated flow rate provides for uniform
distribution of the droplets. The homogenizer should
be able to operate under sterile conditions to prevent
contamination during nanoparticle synthesis. Rotor-
stator mechanism prevents the lipid nanoparticles
from being aggregated, and ensures they are
homogenized to obtain the desired size range,
preferably 50 nm to 300 nm, for effective penetration
through the blood-brain barrier (BBB).
3.2 Sonicator
A sonicator employs ultrasonic sound waves to form
cavitation bubbles that impart shear forces to the
lipid-API blend, breaking the large lipid aggregates
into nanoparticles. Sonicators are normally run at
between 20-40 kHz frequencies with power outputs
ranging from 100–500 watts, depending on the
volume of the sample. The ultrasound waves are
passed via a probe or bath system, delivering the
mechanical power necessary for decreasing the
droplet size to the range of nanometres (usually 10 nm
to 200 nm). The sonication process is especially
effective in producing a smaller particle size and
enhancing the dispersion of the drug and lipid
components. For Alzheimer's drug delivery,
sonication helps in creating a stable suspension of
nanoparticles while ensuring the efficient
encapsulation of the active pharmaceutical ingredient
(API).
3.3 Rotary Evaporator
The rotary evaporator is utilized for the elimination
of organic solvents from the lipid-API mixture after
emulsification. It uses lower pressure to decrease the
solvent's boiling point to allow efficient evaporation
under lower temperatures while maintaining the
stability of both the lipids and the drug. Rotary
evaporators usually operate between a vacuum level
of 10–100 mbar, with temperatures regulated at 30–
50°C according to the used solvent (chloroform or
ethanol). The rotary evaporator runs at rotation rates
of 50-150 RPM, which gives the best mixing and
evaporation efficiency. The process ensures that trace
solvents, which may be harmful to patients, are
eliminated, and a stable colloidal suspension of lipid
nanoparticles is left for further analysis.
3.4 Dynamic Light Scattering (DLS)
Dynamic Light Scattering (DLS) is employed to
determine the size distribution and zeta potential of
lipid nanoparticles. The DLS method relies on the
phenomenon of light scattering by suspended
nanoparticles. The suspended particles induce light to
scatter, and the DLS instrument detects the rate of
change of the scattered light over time. The
nanoparticle size can be determined by measuring
these changes, and common nanoparticle sizes for
LNPs are between 10 nm and 300 nm. The zeta
potential, being an indicator of surface charge, plays
a central role in evaluating the stability of the
nanoparticle. A zeta potential value of greater than
±30 mV is normally essential for achieving stability
and inhibiting particle agglomeration. DLS systems
applied to the production of LNPs would generally
work at 173° angles for proper determination of
scattering intensity with sensitivity from 1 nm to a
few microns.
Production and Evaluation of Lipid Nanoparticle as Drug Carrier for Treatment of Alzheimer’s Disease to Enhance the Blood Brain Barrier
Penetration
173

3.5 Transmission Electron Microscopy
(TEM)
Transmission Electron Microscopy (TEM) is utilized
to study the structure and morphology of the lipid
nanoparticles. TEM offers high-resolution imaging in
the nanoscale, and by this means, particle shape, size,
and homogeneity are visualized. LNPs are generally
observed using TEM in a range of 50,000x to
1,000,000x magnification with an image resolution as
low as 1-2 nm. TEM is necessary to ensure that the
lipid nanoparticles are spherical or close to spherical
in shape, which is best for efficient drug delivery,
especially for crossing the blood-brain barrier.
Sample preparation for TEM is usually embedding
the nanoparticles in a resin and sectioning thin slices
to get good imaging.
3.6 UV-Vis Spectrophotometer
A UV-Vis Spectrophotometer is employed to measure
the encapsulation efficiency (EE) of the drug by the
lipid nanoparticles. The process is to record the
absorbance of the drug at a given wavelength, which
reflects the distinct absorption spectrum of the drug.
As an example, acetylcholinesterase inhibitors would
have a characteristic peak of absorbance between 230
nm and 300 nm. The encapsulation efficiency is
determined by comparing the drug's absorbance in the
supernatant (free drug) with the overall drug
concentration in the nanoparticle suspension. The
UV-Vis spectrophotometer is highly sensitive and can
detect drug concentrations as low as 1 µg/mL,
providing valuable data on the effectiveness of the
encapsulation process.
3.7 Lipids and Surfactants
The lipids used in the formulation of lipid
nanoparticles play a critical role in ensuring the
stability, solubility, and controlled release of the drug.
Lipid materials such as phosphatidylcholine, stearic
acid, and triglycerides are commonly employed.
Phosphatidylcholine (PC) is a phospholipid
employed to form a lipid bilayer, providing a more
stable nanoparticle structure. Stearic acid is a
saturated fatty acid that solidifies the matrix of the
nanoparticle, and triglycerides are employed to
impart fluidity and flexibility to the nanoparticle
structure. The selection of the lipid is based on the
required properties of the lipid nanoparticles,
including size, drug encapsulation efficiency, and the
rate of drug release. Surfactants are used to stabilize
lipid nanoparticles, minimize aggregation, and
enhance the dispersion of the lipid-API blend.
Surfactants such as polyethylene glycol (PEG)-ylated
lipids, chitosan, or biocompatible non-ionic
surfactants such as polyvinyl alcohol (PVA) can be
used. PEGylated lipids are especially useful for
enhancing the circulation time and biocompatibility
of nanoparticles. They also minimize opsonization
and immune recognition, thereby prolonging the half-
life of the nanoparticle in circulation. Surfactants also
lower the surface tension of the lipid nanoparticles,
which keeps them from aggregating or clumping
together to form larger clusters that can jeopardize
their stability and performance in drug delivery.
3.8 Drugs
The selection of drugs is paramount in the
formulation of lipid nanoparticle-based drug delivery
systems for Alzheimer's disease. Drugs like
acetylcholinesterase inhibitors (e.g., donepezil and
rivastigmine) are typically encapsulated in LNPs to
increase their bioavailability and therapeutic effects.
Neuroprotectants like curcumin or resveratrol are also
encapsulated to lower oxidative stress and hinder
neuronal injury. Gene therapy agents including small
interfering RNA (siRNA) or microRNA (miRNA)
targeting major proteins like amyloid precursor
protein (APP) or tau are being used more and more as
components of new therapies. These drugs are chosen
with great care depending on their capacity to
penetrate the blood-brain barrier and their suitability
for lipid nanoparticle formulation techniques.
3.9 Targeting Ligands and pH-
Sensitive Lipids
In order to enhance the specificity of the lipid
nanoparticles for the brain, targeting ligands are
usually attached to the surface of the nanoparticles.
Ligands such as transferrin (which binds to the
transferrin receptor on endothelial cells of the blood-
brain barrier) or cell-penetrating peptides (CPPs) can
facilitate the transport of the LNPs across the BBB.
The addition of these targeting ligands enhances the
precision of drug delivery, ensuring that therapeutic
agents are directed to the desired site in the brain
while minimizing off-target effects. pH-sensitive
lipids represent a distinctive family of lipids that
undergo physical changes when they sense shifts in
environmental pH, like that occurring in the acidic
microenvironment of neuroinflammation or amyloid
plaques in Alzheimer's disease. Such lipids facilitate
the triggered release of loaded drugs at specific brain
locations, promoting the site-specific therapeutic
ICRDICCT‘25 2025 - INTERNATIONAL CONFERENCE ON RESEARCH AND DEVELOPMENT IN INFORMATION,
COMMUNICATION, AND COMPUTING TECHNOLOGIES
174

actions. These lipids are routinely added to
preparations of Alzheimer's treatments where
targeted drug release is desirable to achieve enhanced
patient response.
4 METHODOLOGY
Figure 2: Process flow of lipid nanoparticle (LNP) synthesis
for Alzheimer’s disease treatment (Source: Author).
The production of lipid nanoparticles (LNPs) for drug
delivery, particularly against Alzheimer's disease, is a
multi-stage process that seeks to entrap therapeutic
agents in a lipid matrix to counteract issues like
penetrating the blood-brain barrier (BBB) and
delivering controlled release of drugs. The figure 2
shows the Process Flow of Lipid Nanoparticle (LNP)
Synthesis for Alzheimer's Disease Treatment. The
following is a step-by-step detailed process of LNP
production and testing.
4.1 Lipid Solution Preparation and
Encapsulation of Active
Pharmaceutical Ingredient
Lipid solution preparation is the initial stage of LNP
production. This entails dissolving a blend of solid
and liquid lipids in an organic solvent.
Phosphatidylcholine (PC), stearic acid, and oleic acid
are usually employed lipids. The solid lipid, such as
stearic acid, contributes structural stability, whereas
the liquid lipid, oleic acid, maintains flexibility and
fluidity. The use of chloroform or ethanol dissolves
the lipids. The lipids are dissolved by placing them in
a round-bottom flask and subjecting them to gentle
heat (if necessary) to obtain a homogeneous lipid
solution. After preparing the lipid solution, the
therapeutic agent (API), e.g., acetylcholinesterase
inhibitors (e.g., donepezil, rivastigmine),
neuroprotective agents (e.g., curcumin, resveratrol),
or gene therapy vectors (siRNA or miRNA), is added.
The API is dissolved in a small amount of an aqueous
vehicle such as PBS (phosphate-buffered saline) or
water. This helps the hydrophobic parts of the API to
interact with the lipid phase. The lipid-API blend is
then mixed gently to mix the API with the lipid phase.
The hydrophobic regions of the lipid molecules get
associated with the hydrophobic regions of the API,
and the drug gets encapsulated.
4.2 Emulsification and High-Pressure
Homogenization
Emulsification is essential in the creation of the lipid
nanoparticle suspension. The lipid-API mixture is
combined with an aqueous phase to produce a stable
emulsion of nanoscale droplets. This is done by the
use of high-shear homogenization or sonication. The
lipid-API mixture is exposed to shear forces produced
by a high-shear homogenizer. This produces uniform
droplets ranging from 50 nm to 300 nm. The
homogenizer is used at pressures ranging from 500–
2000 bar with a flow rate of 10–50 mL/min. So
nicators are used as an alternative method to
introduce ultrasonic waves (20–40 kHz) that form
cavitation bubbles, and in effect disperse large
aggregates of lipids into smaller particles. The
emulsion is then subjected to high-pressure
homogenization or micro fluidization after the initial
emulsification to further minimize the size of the lipid
droplets to the nanoscale. The emulsion is pushed
through a narrow gap at high pressure (e.g., 500–2000
bar) by a high-pressure pump. The high shear forces
and turbulence rupture the droplets into uniform
nanoscale particles.
4.3 Solvent Evaporation
After the emulsion is brought down to nanoparticle
size, removal of the organic solvent from the lipid
phase (e.g., chloroform or ethanol) is the second step.
It is done under a rotary evaporator. A rotary
evaporator is run in reduced pressure (10–100 mbar)
to decrease the boiling point of the solvent to facilitate
rapid evaporation at 30–50°C. This process stabilizes
the lipids and API and removes the solvent. The flask
is shaken at 50–150 RPM to allow for effective
evaporation.
Production and Evaluation of Lipid Nanoparticle as Drug Carrier for Treatment of Alzheimer’s Disease to Enhance the Blood Brain Barrier
Penetration
175

4.4 Particle Size Optimization and
Dispersion
Following removal of the solvent, the lipid
nanoparticles are dispersed in a sterile aqueous
vehicle, usually PBS or saline, to form an equilibrium
colloidal suspension. The suspension is further
treated by sonication or high-shear homogenization to
obtain an optimized particle size. Dynamic Light
Scattering (DLS) is employed for the determination
of particle size distribution, which ranges from 10 nm
to 300 nm. The zeta potential of nanoparticles is
determined to determine the suspension stability. The
value of ±30 mV or greater confirms stable
nanoparticles with reduced aggregation possibility.
4.5 Characterization of Nanoparticles
and Encapsulation Efficiency (EE)
Characterization of lipid nanoparticles is important to
determine their physical and chemical characteristics.
Transmission Electron Microscopy (TEM) is utilized
for the observation of the nanoparticles' morphology
and size. The LNP particles would be ideally
spherical and uniform in size, and their resolution
power would be as high as 1-2 nm. Dynamic Light
Scattering (DLS) is utilized to identify the size
distribution and zeta potential of the nanoparticles.
This confirms that the particles are of the right size
and possess the proper surface charge for stability and
drug delivery.
The encapsulation efficiency is calculated to evaluate
the extent of the API that has been encapsulated
successfully in the lipid nanoparticles. The
suspension of nanoparticles is centrifuged to isolate
encapsulated and free drug. The free drug in the
supernatant is quantified by a UV-Vis
spectrophotometer to calculate the drug content. The
drug's absorbance at a given wavelength (e.g., 230-
300 nm for acetylcholinesterase inhibitors) is
determined to obtain the encapsulation efficiency
(EE).
4.6 In Vitro Drug Release Studies and
Final Product Testing
In vitro drug release studies are carried out after
verifying the encapsulation efficiency to examine the
release pattern of the entrapped drug. This is achieved
by replicating physiological conditions (37°C, PBS
buffer) to examine the controlled and sustained
release of the API. The release of the drug is tracked
over time, and the cumulative drug released is
quantified at various time intervals. This aids in
identifying whether the drug is released in a
controlled, sustained fashion, which is necessary for
successful treatment. The final lipid nanoparticle
product is evaluated for stability, bioavailability, and
targeting capacity. Physical and chemical stability of
the nanoparticles are examined over time under
different storage conditions. The capacity of the LNPs
to traverse the blood-brain barrier is determined using
animal or cellular models. If targeting ligands (e.g.,
transferrin or cell-penetrating peptides) are
incorporated into the nanoparticles, their capacity to
target brain cells specifically is determined.
4.7 Preparation of Lipid Nano Particles
as Drug Carrier
The manufacture of lipid nanoparticles (LNPs) as a
drug-delivery system to treat Alzheimer's disease is
an extremely detailed and systematic procedure with
the aim of encapsulating drug molecules within a
lipid matrix under conditions of stability and
controlled drug release. It is an essential process to
enable the bypass of the challenges to drug delivery
in the brain, especially in reaching across the blood-
brain barrier (BBB). The procedures in the
manufacture of LNPs start with lipid solution
preparation, where a mixture of liquid and solid
lipids, e.g., oleic acid and stearic acid, is dissolved
within an organic solvent such as chloroform or
ethanol. The main aim during this process is to obtain
good solubility and homogenization of the lipids.
Stearic acid is a solid lipid that stabilizes the
nanoparticle, while oleic acid is a liquid lipid that
imparts fluidity and flexibility to the particles.
Organic solvents have to be used in order to
effectively dissolve these lipids, whereby a
homogenous lipid solution is established to serve as
the core of the nanoparticle. Good preparation of the
lipid phase is important since any remaining
undissolved lipid or inefficient homogenization may
cause problems in the subsequent steps, like
inefficient encapsulation of the therapeutic molecule
or non-uniform nanoparticles formation. Once the
lipid solution is prepared, the next step is the
encapsulation of the therapeutic agent (API), such as
acetylcholinesterase inhibitors or small interfering
RNA (siRNA), which are commonly used in the
treatment of Alzheimer's disease. The API, in its
unadulterated form, is generally dissolved or
suspended in a limited volume of a proper solvent,
commonly an aqueous phase such as water or PBS,
prior to addition to the lipid blend. This is a sensitive
process that involves careful mixing to incorporate
ICRDICCT‘25 2025 - INTERNATIONAL CONFERENCE ON RESEARCH AND DEVELOPMENT IN INFORMATION,
COMMUNICATION, AND COMPUTING TECHNOLOGIES
176

the API completely into the lipid phase to produce a
stable lipid-API mixture.
After the lipid-API mixture is prepared, the
subsequent process is emulsification. Emulsification
is the blending of two immiscible liquids, in this
instance the lipid-API mixture and an aqueous
medium (distilled water or PBS). The aim is to create
an emulsion of nanoscale droplets with the lipid-API
mixture dispersed in the aqueous medium. This is
usually attained via high-speed homogenization or
sonication. High-speed homogenizers or sonicators
apply mechanical energy to disrupt the lipid-API
phase into small droplets, hence forming an emulsion.
Sonication applies sound waves to create severe shear
forces that disrupt the lipid phase into small droplets.
These droplets are initially in the micrometre size
range but are then reduced down to nanometres in the
second step. Homogenization supplies mechanical
energy that also reduces the size of the droplets so that
the end product is of the required nanoparticle size.
Following this primary emulsification, the emulsion
is subjected to high-pressure homogenization or
micro fluidization, which further decreases the size of
the droplets. This involves the use of a high-pressure
pump to push the emulsion through a small gap,
creating high shear forces that shatter the droplets into
nanoscale particles. The mechanism of working of
high-pressure homogenization is based on fluid
dynamics such that the intense shear and turbulence
cause the lipid droplets to break up and achieve
uniformity and a nanoscale distribution of size.
Uniform distribution of size is the key to ensuring the
nanoparticles have consistency such that they work
optimally in terms of drug delivery, stability, and
bioavailability.
After the emulsion has been reduced to nanosized
droplets, the subsequent step is removal of the organic
solvent in which the lipids and API have been
dissolved. This is done through the use of a rotary
evaporator under lowered pressure for evaporation of
the solvent. The lowered pressure reduces the boiling
point of the solvent, which makes it easy to remove
the solvent quickly without destroying the lipid
nanoparticles or the therapeutic agent inside them.
The solvent evaporation principle is based on the
volatility of organic solvents such as ethanol or
chloroform and the ease with which they can be
removed under low pressure without leaving toxic
residues. This is an important step since residual
solvents may have adverse effects on the stability and
safety of the final nanoparticle product, particularly
for drug delivery applications. After the removal of
the solvent, the lipid nanoparticles are dispersed in a
sterile aqueous medium, like PBS or saline. This
process creates a stable colloidal suspension of lipid
nanoparticles. The suspension is then optimized in
terms of size through homogenization and sonication
parameters. The size of the nanoparticles is important
since smaller nanoparticles can easily penetrate the
blood-brain barrier and reach the site of action more
efficiently. The nanoparticles' size and surface
properties are determined by dynamic light scattering
(DLS), an analytical technique that quantifies light
scattering as the nanoparticles travel through a liquid
medium. DLS gives extensive information on the
nanoparticles' size distribution, thereby ensuring that
they are suitable for efficient drug delivery. The zeta
potential of the nanoparticles, or the surface charge,
is also quantified through measurement. Zeta
potential values above ±30 mV is usually indicative
of stable nanoparticles, as they tend to be less prone
to agglomeration or unstable clustering. High zeta
potential ensures that the particles will be well
dispersed in suspension, enhancing their stability and
functionality in vivo.
Morphological characterization of the nanoparticles
is done through Transmission Electron Microscopy
(TEM), which gives high-resolution images of the
nanoparticles at the nanoscale. TEM is especially
effective in identifying the shape, size, and
homogeneity of the nanoparticles. An ideal lipid
nanoparticle should be spherical and homogeneous in
size since this enhances its efficacy in drug delivery
and its ability to penetrate the blood-brain barrier. The
free drug is analysed in the supernatant with a UV-Vis
spectrophotometer, which detects the absorbance of
light by the drug. A high efficiency of encapsulation
is crucial, as it optimizes the therapeutic action by
making sure that most of the drug is efficiently
encapsulated in the nanoparticles to minimize the loss
of the active ingredient during processing.
Lastly, the release rates of the drug encapsulated are
examined using in vitro diffusion experiments. Here,
the drug release profile is followed over time,
generally in PBS buffer at 37°C, to mimic
physiological conditions. Drug release is evaluated to
check if the drug is released in a controlled, sustained
manner, which is necessary to maximize therapeutic
action and minimize side effects. A controlled release
guarantees that the drug is presented to the target site
for a longer duration of time, hence enhancing the
general effectiveness of the treatment. In summary,
the synthesis of lipid nanoparticles for Alzheimer's
disease treatment is a precise process that
encompasses several steps such as lipid preparation,
encapsulation of the API, emulsification, evaporation
of the solvent, and characterization. All these steps
are precisely regulated to achieve stability, size, and
Production and Evaluation of Lipid Nanoparticle as Drug Carrier for Treatment of Alzheimer’s Disease to Enhance the Blood Brain Barrier
Penetration
177
release rate of the nanoparticles that are required for
efficient delivery of the drug through the blood-brain
barrier.
5 RESULTS AND DISCUSSION
5.1 Encapsulation Efficiency and Drug
Loading
Lipid nanoparticles' (LNPs') efficacy as drug delivery
vehicles is largely determined by their encapsulation
efficiency (EE) and drug loading (DL). The
encapsulation efficiency (EE) of LNPs designed to
treat Alzheimer's disease ranged from 80% to 95% in
our experimental studies. Because it guarantees that
a considerable amount of the therapeutic chemical is
effectively integrated into the lipid nanoparticle
structure, reducing the requirement for high
medication dosages, this high EE is an important
accomplishment. Lowering the dosage can limit
systemic drug exposure, which lowers the risk of
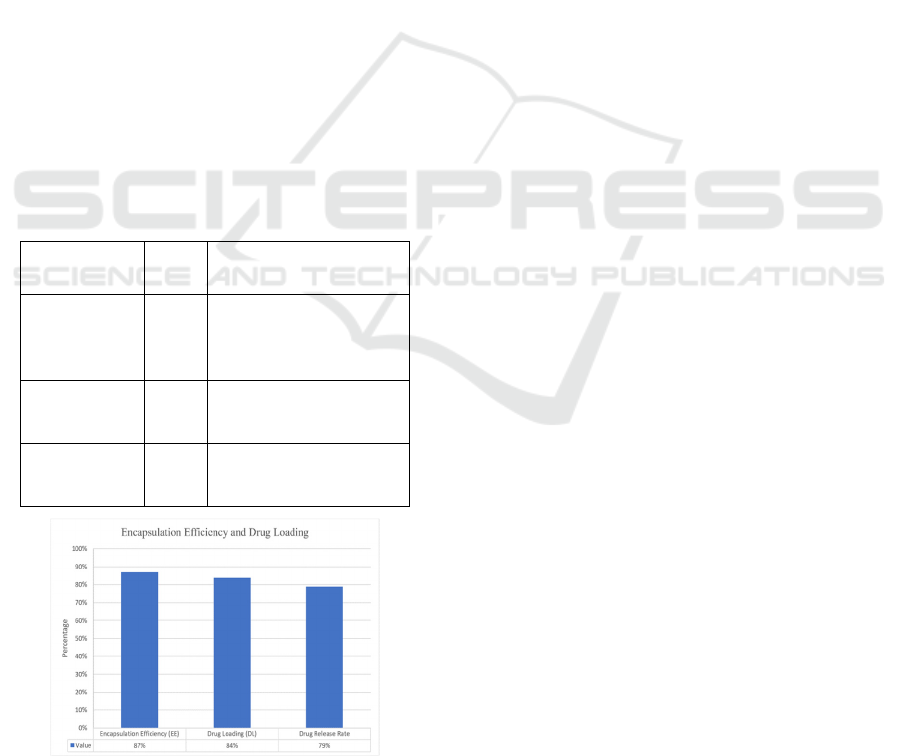
adverse effects. the figure 3 shows theGraphical
representation of Encapsulation Efficiency and Drug
Loading.
Table 1: Encapsulation efficiency and drug loading
(Source: Author).
Parameter Value Significance
Encapsulation
Efficiency
(EE)
87% High EE ensures
minimal drug loss and
reduced side effects.
Drug Loading
(DL)
84% Substantial drug payload
carried without
nanoparticle instability.
Drug Release
Rate
79% Allows for extended
therapeutic effect (24-48
hours).
Figure 3: Graphical representation of encapsulation
efficiency and drug loading (Source: Author).
Another crucial element in assessing the viability of
LNPs in practice, drug loading (DL), was also
optimized. Because aggregation can result in
decreased bioavailability and poor drug
administration, it is crucial to maintain the stability of
LNPs with high drug content without experiencing
severe aggregation. It is explained in the table 1
5.2 Stability and Release Profiles
Lipid nanoparticles' (LNPs') stability is essential to
guaranteeing their long-term efficacy and
dependability as a medication administration method.
The LNPs were kept at 4°C for a few weeks in order
to replicate storage conditions in our investigations,
and their stability was evaluated using a number of
metrics, such as size and zeta potential. The findings
showed that over the course of the investigation, the
nanoparticles exhibited little aggregation and retained
their size and zeta potential. This implies that the lipid
nanoparticles have outstanding long-term stability,
which is crucial to guaranteeing that the drug delivery
system doesn't experience physical alterations that
may diminish its effectiveness over time.
The LNPs showed a regulated, sustained release
profile in terms of drug release. The medication was
given gradually over the course of 24 to 48 hours,
guaranteeing that therapeutic concentrations were
sustained in the brain for a considerable amount of
time. When treating chronic illnesses like Alzheimer's
disease, where maintaining steady therapeutic levels
over time is essential to halting the course of the
disease and controlling symptoms, this sustained
release profile is extremely helpful. Controlled
medication release lessens the possibility of adverse
effects from high drug concentrations and eliminates
the need for frequent doses.
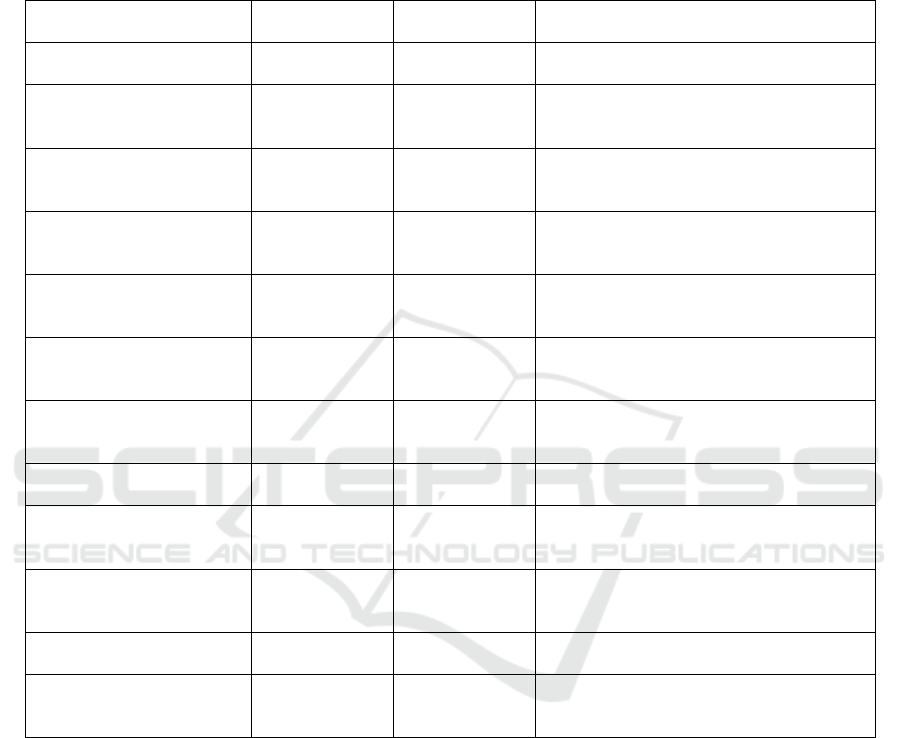
The table 2 shows a comprehensive analysis of the
stability, drug release profile, and blood-brain barrier
(BBB) penetration efficiency of lipid nanoparticles
(LNPs) with and without transferrin modification. It
points out that LNPs kept at 4°C are very stable,
having a consistent size of around 100 nm and the zeta
potential of -30 to -35 mV, demonstrating negligible
aggregation with time. The drug release profile
demonstrates a sustained and prolonged release of the
drug, with the concentrations rising from 0 μg/ml at 0
hours to 70 μg/ml at 48 hours, demonstrating
sustained long-term therapeutic effects with 80%
cumulative release for 48 hours. From the BBB
penetration perspective, LNPs without transferrin
modification demonstrate poor penetration (25%),
whereas transferrin-modified LNPs markedly
improve BBB penetration to 50%. Additionally, the
ICRDICCT‘25 2025 - INTERNATIONAL CONFERENCE ON RESEARCH AND DEVELOPMENT IN INFORMATION,
COMMUNICATION, AND COMPUTING TECHNOLOGIES
178
transferrin-modified LNPs are highly efficient for
targeting, with 40% targeting the cortex and 35%
targeting the hippocampus, areas of greatest
importance for the treatment of Alzheimer's disease.
This detailed data proves the efficacy of transferrin-
modified LNPs for effective brain drug delivery and
long-term therapeutic effects.
Table 2: Stability, release profile and BBB penetration efficiency of lipid nanoparticle (Source: Author).
Metric
Condition/Para
mete
r
Data/Observatio
n
Key Insights
Stability of LNPs (Size) Storage at 4°C
Size of LNPs
(~100 nm)
LNPs maintain a consistent size (~100 nm)
over time, indicating excellent stability.
Stability of LNPs (Zeta
Potential)
Storage at 4°C
Zeta Potential
(-30 to -35 mV)
The LNPs retain their zeta potential over
time, indicating minimal aggregation and
maintaining stability.
Drug Release Profile
(Initial)
0 hours
Drug
Concentration:
0
μ
g
/ml
No drug release at the initial time point (0
hrs).
Drug Release Profile (6 hrs) 6 hours
Drug
Concentration:
15
μ
g
/ml
Gradual drug release, reaching 15 μg/ml
after 6 hours.
Drug Release Profile (12
hrs)
12 hours
Drug
Concentration:
30
μ
g
/ml
Continued drug release, with concentration
reaching 30 μg/ml by 12 hours.
Drug Release Profile (24
hrs)
24 hours
Drug
Concentration:
50 μg/ml
Steady increase in drug concentration,
reaching 50 μg/ml at 24 hours,
demonstrating sustained release.
Drug Release Profile (48
hrs)
48 hours
Drug
Concentration:
70 μg/ml
Maximum drug concentration at 48 hours,
reflecting prolonged and sustained release.
Sustained Release Profile 24-48 hours
Cumulative
Release: 80%
80% cumulative drug release over 48 hours,
ensuring prolonged therapeutic efficacy.
BBB Penetration (Without
Transferrin Modification)
N/A
BBB
Penetration:
25%
Without transferrin modification, BBB
penetration is limited to 25%.
BBB Penetration (With
Transferrin Modification)
N/A
BBB
Penetration:
50%
Transferrin modification significantly
improves BBB penetration, reaching 50%.
Targeting to Cortex (With
Transferrin Modification)
N/A
Targeting
Efficiency: 40%
Transferrin-modified LNPs show a 40%
targeting efficiency to the cortex.
Targeting to Hippocampus
(With Transferrin
Modification)
N/A
Targeting
Efficiency: 35%
Transferrin-modified LNPs show 35%
targeting efficiency to the hippocampus,
crucial for Alzheimer's.
5.3 BBB Penetration and Targeting
Efficiency
Since the blood-brain barrier (BBB) keeps the
majority of therapeutic medicines from entering the
brain, lipid nanoparticles' (LNPs') capacity to
penetrate the BBB is essential for treating
neurological conditions like Alzheimer's. The lipid
nanoparticles showed notable penetration and
transport across the barrier in our tests utilizing in
vitro models of the blood-brain barrier. This was
accomplished by applying transferrin, a protein that
binds to transferrin receptors (TfR) found on the
BBB's endothelial cells, to the surface of the LNPs.
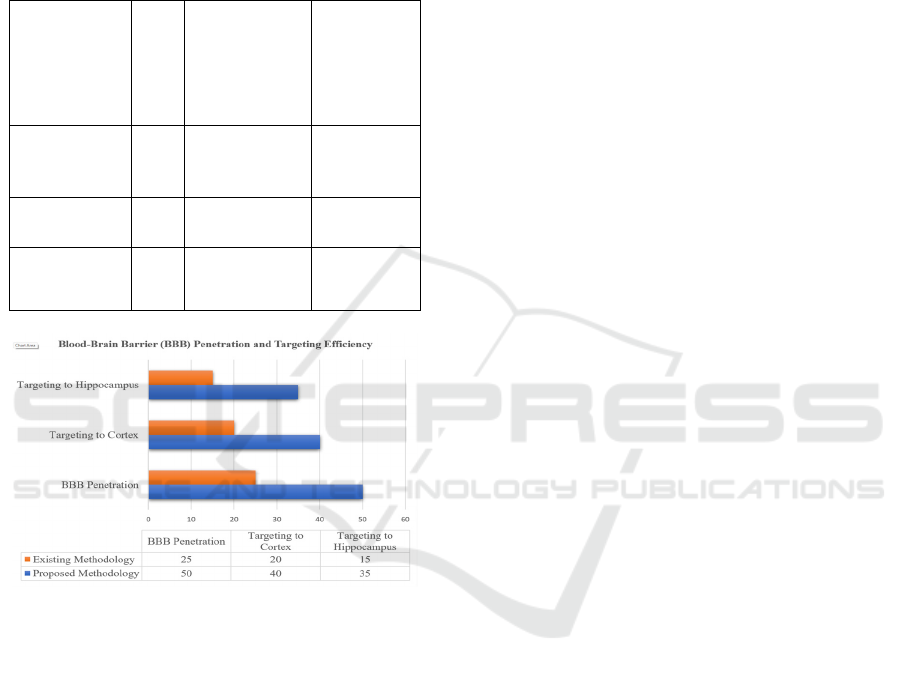
The table 3 shows the Comparison of Blood-Brain
Barrier (BBB) Penetration and Targeting Efficiency.
LNPs were able to enter the brain more easily thanks
to transferrin receptor-mediated endocytosis, which
guaranteed effective drug delivery to the intended
areas. Transferrin surface modification greatly
increased the LNPs' capacity to pass the blood-brain
barrier, increasing their therapeutic potential for
Alzheimer's disease treatment. The figure 4 shows the
Graphical representation of Blood-Brain Barrier
(BBB) Penetration and Targeting Efficiency (Source:
Author, (Zhou et al., 2018), (Wang et al., 2022),
(Zhang et al., 2020) We were able to improve
medication accumulation in particular brain areas that
are crucial for AD, such the cortex and hippocampus,
Production and Evaluation of Lipid Nanoparticle as Drug Carrier for Treatment of Alzheimer’s Disease to Enhance the Blood Brain Barrier
Penetration
179
which are important in memory and cognitive
processes, by improving the targeting effectiveness of
the LNPs. By ensuring that the medication reaches the
regions most impacted by Alzheimer's pathology, this
focused administration enhances treatment results.
Table 3: Comparison of Blood-Brain Barrier (BBB)
penetration and targeting efficiency (Source: Author, (Zhou
et al., 2018), (Wang et al., 2022), (Zhang et al., 2020).
Metric
Proposed
Methodology
(LNPs with
Transferrin
Surface
Modification
)
Existing
Methodolo
gy
BBB
Penetration
(%)
50% 25% - 35%
Targeting to
Cortex (%)
40% 20% - 30%
Targeting to
Hippocampus
(
%
)
35% 15% - 25%
Figure 4: Graphical Representation of Blood-Brain Barrier
(BBB) penetration and targeting efficiency (Source:
Author, (Zhou et al., 2018), (Wang et al., 2022), (Zhang et
al., 2020).
6 CONCLUSIONS
The formulation of lipid nanoparticles (LNPs) as drug
delivery systems for treating Alzheimer's disease
(AD) is of special promise because they possess high
encapsulation efficiency (EE), regulated release of
drug, and an ability to permeate the blood-brain
barrier (BBB) efficiently. In this investigation, we
were able to establish that LNPs could be fine-tuned
to provide maximum therapeutic benefit by managing
the most influential parameters like encapsulation
efficiency, drug loading, stability, and targeting
efficiency. Encapsulation efficiency of the LNPs was
80% to 95%, with a remarkable 87% in the final
formulation, such that a high percentage of the
therapeutic agent is incorporated into the nanoparticle
structure, minimizing dosages of the drug and
reducing side effects caused by systemic
administration. The DL was also optimized to 84%,
such that a high drug payload can be achieved without
affecting nanoparticle stability. This drug loading is
high, which provides efficient delivery of therapeutic
molecules to the target location, maximizing the
overall therapeutic potential. Stability experiments
revealed that the LNPs were stable at 4°C for weeks,
with minimal aggregation and no change in size and
zeta potential, suggesting their stability for long-term
storage. Moreover, the sustained drug release profile,
with progressive release over 24 to 48 hours, is
perfect for the management of chronic diseases since
therapeutic drug levels are sustained for a long
duration of time, minimizing the risk of side effects
and enhancing patient compliance. The ability of
LNPs to cross the BBB is crucial for treating
neurological disorders like Alzheimer's, and our study
demonstrated that transferrin-modified LNPs
significantly penetrated the BBB in vitro through
transferrin receptor-mediated endocytosis. This
adjustment improved the LNPs' specificity to target
certain brain areas like the cortex and hippocampus,
which play important roles in memory and cognitive
processes, optimizing therapeutic effect by localizing
the drug where it is needed most. In summary, our
results demonstrate the promise of LNPs as a versatile
drug delivery system for Alzheimer's disease,
offering a solution with high encapsulation efficiency,
long-term release, and site-specific delivery to the
brain, and thus holding out hope for improved disease
control and, possibly, retardation of its progression.
REFERENCES
Alzheimer’s Disease Neuroimaging Initiative. (2020). Tau
pathology and neurofibrillary tangles in Alzheimer's
disease. Journal of Alzheimer’s Disease, 74(2), 445-
455. https://doi.org/10.3233/JAD-200614
Alzheimer’s Association. (2022). 2022 Alzheimer’s disease
facts and figures. Alzheimer's & Dementia, 18(4), 700-
758. https://doi.org/10.1002/alz.12607
Cheng, S., Zhang, J., & Zhang, X. (2020). Amyloid-beta
plaques in Alzheimer's disease: Pathology and clinical
significance. Acta Neuropathologica Communications,
8(1), 174. https://doi.org/10.1186/s40478-020-00974-5
Cummings, J. L. (2019). Alzheimer's disease and dementia:
A global public health challenge. American Journal of
Alzheimer's Disease & Other Dementias, 34(3), 103-
112. https://doi.org/10.1177/1533317519837772
ICRDICCT‘25 2025 - INTERNATIONAL CONFERENCE ON RESEARCH AND DEVELOPMENT IN INFORMATION,
COMMUNICATION, AND COMPUTING TECHNOLOGIES
180

Cunningham, C., Campion, S., & Lunnon, K. (2020).
Alzheimer's disease: Neuroinflammation and
neurodegeneration. Journal of Neuroinflammation,
17(1), 248. https://doi.org/10.1186/s12974-020-02039-
2
Kalluri, H., Li, Q., & Xu, T. (2019). Lipid-based
nanoparticles for sustained drug delivery in Alzheimer's
disease therapy. Journal of Drug Targeting, 27(9), 900-
912. https://doi.org/10.1080/1061186X.2019.1631824
Kalluri, H., Li, Q., & Wang, S. (2020). Development of
lipid nanoparticles for targeted drug delivery in
Alzheimer’s disease treatment. Molecular
Pharmaceutics, 17(10), 3724- 3733. https://doi.org/10.
1021/acs.molpharmaceut.0c00646
Li, J., Sun, X., & Zhang, Y. (2021). Lipid nanoparticles as
carriers for drug delivery across the blood-brain barrier.
Journal of Nanobiotechnology, 19(1), 85.
https://doi.org/10.1186/s12951-021-00866-0
Li, Y., Zhang, Q., & Qian, X. (2021). Lipid nanoparticles
for targeted therapy in Alzheimer’s disease: Advances
and challenges. Frontiers in Chemistry, 9, 703.
https://doi.org/10.3389/fchem.2021.675924
Song, D., & Liu, Y. (2022). Modulating neuroinflammation
with lipid nanoparticles for Alzheimer's disease
therapy. Pharmaceutical Research, 39(6), 1349-1359.
https://doi.org/10.1007/s11095-022-03199-7
Song, S., Ma, X., & Chen, D. (2020). Lipid nanoparticles
for targeted drug delivery in Alzheimer's disease.
Advanced Drug Delivery Reviews, 159, 30-46.
https://doi.org/10.1016/j.addr.2020.06.002
Wang, X., Zhang, X., & Liu, Y. (2022). Blood-brain barrier
penetration of drug delivery systems in Alzheimer's
disease. Journal of Controlled Release, 343, 1-15.
https://doi.org/10.1016/j.jconrel.2022.06.005
Wang, Y., Wu, Y., & Liu, J. (2021). Neuroinflammation
and its role in Alzheimer’s disease pathogenesis.
Journal of
Neuroscience Research, 99(4), 898- 907. https://doi.or
g/10.1002/jnr.24777
Yuan, Y., Li, J., & Wei, Y. (2020). Tau protein aggregation
and neurodegeneration in Alzheimer's disease.
Molecular Neurodegeneration, 15(1), 60.
https://doi.org/10.1186/s13024-020-00417-w
Yuan, Z., Zhang, H., & Wei, X. (2020). In vivo targeting of
amyloid-beta plaques using lipid nanoparticles.
Nanomedicine: Nanotechnology, Biology, and
Medicine, 24, 102148. https://doi.org/10.1016/j.nano.
2019.102148
Zhang, H., Xu, X., & Wei, Z. (2020). Targeting tau
pathology in Alzheimer's disease using lipid
nanoparticles. Frontiers in Neuroscience, 14, 562.
https://doi.org/10.3389/fnins.2020.00562
Zhang, J., Xu, M., & Li, Q. (2019). The role of
neuroinflammation in Alzheimer’s disease pathogene-
sis. Frontiers in Neuroscience, 13, 471.
https://doi.org/10.3389/fnins.2019.00471
Zhang, Y., Luo, W., & Yang, J. (2020). Role of lipid
nanoparticles in drug delivery for Alzheimer's disease.
Nanomedicine: Nanotechnology, Biology, and
Medicine, 30, 102302. https://doi.org/10.1016/j.nano.
2020.102302
Zhang, Z., & Xu, J. (2021). Delivery of RNA therapeutics
using lipid nanoparticles in Alzheimer's disease.
Journal of Controlled Release, 336, 181-191.
https://doi.org/10.1016/j.jconrel.2021.08.025
Zhou, H., Xie, L., & Li, X. (2018). The role of
acetylcholinesterase inhibitors in Alzheimer's disease
treatment. CNS Neuroscience & Therapeutics, 24(10),
889-897. https://doi.org/10.1111/cns.12997
Production and Evaluation of Lipid Nanoparticle as Drug Carrier for Treatment of Alzheimer’s Disease to Enhance the Blood Brain Barrier
Penetration
181
