
Weakly Supervised Graph Neural Networks for Scalable 3D Phase
Segmentation in Molecular Dynamics Simulations
Abin Shakya
a
and Bijaya B. Karki
b
School of Electrical Engineering and Computer Science, Louisiana State University, U.S.A.
Keywords:
GNN, Phase Segmentation.
Abstract:
Accurate phase identification in large-scale molecular dynamics simulation remains a significant challenge due
to ambiguous boundaries between compositionally distinct regions and the lack of ground truth labels. While
unsupervised methods can perform phase segmentation for small systems through structure-aware segmenta-
tion pipelines, their computational cost becomes prohibitive for large-scale analysis. We present a weakly-
supervised machine learning pipeline that trains Graph Neural Networks (GNNs) to enable scalable phase
segmentation in 3D atomistic systems. Using a physically grounded unsupervised method, we generate weak
labels for small FeMgSiON systems that exhibit Fe-rich (metallic) and Fe-poor (silicate) phase separation.
These labels guide GNNs to learn physically meaningful representations of atomic neighborhoods. Once
trained, the GNNs act as an efficient parametric model, enabling direct segmentation of arbitrarily large atom-
istic systems eliminating the computational overhead of the initial unsupervised pipeline. By learning from
thousands of weakly labeled snapshots, the model discerns latent structural patterns, enhancing both predic-
tion accuracy and generalization to unseen data. This methodology enables efficient, accurate, and physically
consistent phase segmentation in large-scale molecular dynamics, unlocking new possibilities for scalable
analysis in material simulations.
1 INTRODUCTION
Identifying distinct physical or chemical phases is
a fundamental problem in science and engineer-
ing. It enables analysis of phase stability in mate-
rials, core–mantle differentiation in geoscience, and
biomolecular assemblies such as protein condensates
and lipid domains in biology. Across these domains,
accurate phase identification is essential for quantify-
ing composition and tracking interfaces. In molecu-
lar dynamics (MD) simulations, this challenge is of-
ten addressed through semantic segmentation, which
assigns per-element labels to spatial data to iden-
tify physically meaningful regions (Long et al., 2015;
Ronneberger et al., 2015; Chen et al., 2017; Qi et al.,
2017). This approach is especially important for ana-
lyzing complex phenomena such as phase separation,
chemical mixing and interfacial behavior. However,
automated segmentation remains a significant chal-
lenge due to the absence of ground truth labels, the
irregular nature of atomic point clouds, and the pres-
a
https://orcid.org/0009-0000-3176-5629
b
https://orcid.org/0000-0003-2428-0206
ence of nonlinear, diffuse boundaries between phases.
We focus on phase separation in molecular dy-
namics simulations of FeMgSiON, a chemically
complex system representative of bulk earth—under
pressure-temperature conditions of 29 GPa and 3000
K, and 35 GPa and 4000 K (McDonough and Sun,
1995). These simulations produce a series of config-
urations, each capturing the 3D positions of all atoms
within the simulation cell at a given time step. As the
system evolves, it spontaneously separates into chem-
ically distinct regions—most notably a Fe-rich metal-
lic phase and a Fe-poor silicate phase (Shakya et al.,
2024). Although the separation is visually appar-
ent, automating the segmentation of atoms into mean-
ingful phases is non-trivial due to atomic-scale noise
and overlapping compositional transitions. Moreover,
precise boundary identification is essential for accu-
rately evaluating the elemental composition of each
phase, as even minor misclassifications near inter-
faces can significantly skew weight percent estimates.
To address this, we propose a hybrid framework
that combines physics-informed unsupervised analy-
sis with graph-based learning. Our key insight is that
small systems can be segmented using physically mo-
302
Shakya, A. and Karki, B. B.
Weakly Supervised Graph Neural Networks for Scalable 3D Phase Segmentation in Molecular Dynamics Simulations.
DOI: 10.5220/0013709400004000
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 17th International Joint Conference on Knowledge Discovery, Knowledge Engineering and Knowledge Management (IC3K 2025) - Volume 1: KDIR, pages 302-312
Proceedings Copyright © 2025 by SCITEPRESS – Science and Technology Publications, Lda.

tivated heuristics, and the resulting weak labels can
then be used to train graph-based models capable of
generalizing to much larger systems. This approach
bridges the gap between accurate but computationally
expensive unsupervised methods (Lopez et al., 2019)
and scalable predictive models suitable for large-scale
simulations. Our implementation details can be found
here.
1
Our method proceeds in two stages. First, we gen-
erate weak labels for small systems by computing a
smoothed density field of Fe atoms using kernel den-
sity estimation (KDE), followed by K-Means clus-
tering and morphological post-processing (Silverman,
1986; MacQueen, 1967). This unsupervised pipeline
produces per-atom labels for Fe-rich, Fe-poor, and
interfacial boundary regions. Second, we train a
message-passing graph neural network on these weak
labels to learn a mapping from local atomic environ-
ments to phase labels. Atomic snapshots are repre-
sented as graphs, with edges defined by spatial prox-
imity under periodic boundary conditions. We exper-
iment with two distinct GNN architectures—Graph
Convolutional Networks (GCN) and Graph Atten-
tion Networks (GAT)—to evaluate their compatibility
with different message-passing schemes. (Scarselli
et al., 2009; Kipf and Welling, 2017; Hamilton et al.,
2017; Veli
ˇ
ckovi
´
c et al., 2018).
Once trained, the GNN enables fast and accu-
rate segmentation of large-scale atomic systems that
would be prohibitively expensive to label using the
original unsupervised pipeline. For example, the
GNN achieves over two orders of magnitude speedup
on 33,280-atom snapshots compared to the KDE-
based pipeline, while maintaining high fidelity to the
reference labels, especially near phase boundaries.
Moreover, the learned model generalizes to simula-
tions of arbitrary scale without retraining, overcoming
a fundamental limitation of unsupervised approaches.
Beyond this specific application, our work demon-
strates how physically grounded weak supervision
can be used to overcome the dual challenges of la-
bel scarcity and computational scalability in scien-
tific machine learning. While our study targets a spe-
cific problem, the broader strategy—deriving weak
labels from small, well-characterized systems and
training parametric models to generalize segmenta-
tion—applies to a wide range of 3D scientific data in
physics, chemistry, and related domains.
1
Code used in this study available at: https://github.
com/arsenomadridabin/PhaseSegmentationWithGNN
2 RELATED WORK
Phase segmentation in molecular dynamics simula-
tion has been approached through a range of strate-
gies, including geometric heuristics, structure based
classification, statistical binning methods, and data-
driven machine learning techniques. These ap-
proaches differ in how they represent atomic environ-
ments, define boundaries, and balance accuracy with
scalability.
Traditional approaches for identifying phases in
molecular dynamics simulations typically employ
structural or topological heuristics, including Com-
mon Neighbor Analysis (CNA) (Honeycutt and An-
dersen, 1987) and Voronoi-based techniques. CNA
classifies atomic environments by examining the lo-
cal bonding topology—particularly the count and ar-
rangement of shared neighbors—proving particularly
useful for distinguishing crystalline phases and de-
fects. Voronoi-based methods, in contrast, assess co-
ordination environments through geometric tessella-
tion. Stukowski (Stukowski, 2012) offers a detailed
evaluation of these approaches while introducing im-
provements to CNA for multi-phase systems. Al-
though these methods perform well in recognizing
distinct structural patterns, their dependence on rigid
geometric or topological assumptions restricts their
effectiveness in chemically diverse systems, where
phase determination depends more on compositional
variation and gradual transitions rather than clear-cut
symmetry.
Geometric approaches identify phase boundaries
using constructs derived directly from atomic po-
sitions. For instance, alpha shapes (Edelsbrunner
and M
¨
ucke, 1994)—a generalization of convex hulls
(Chazelle, 1993) —have been used to enclose atomic
clusters and define metal-rich regions by drawing a
boundary around Fe atoms (Zhang and Guo, 2009).
Atoms within the alpha shape are classified as metal-
lic, while those outside are treated as silicate. This
method offers an intuitive spatial characterization and
has been used effectively for small, cleanly sepa-
rated systems. However, it assumes sharp, well-
defined boundaries and neglects the diffuse and tran-
sitional nature of phase interfaces often observed
in multi-atom species and high-temperature environ-
ments. The approach also lacks per-atom resolution,
and its reliance on geometric regularity makes it sen-
sitive to noise, parameter tuning, and system size, ul-
timately limiting its applicability to more complex or
disordered atomic configurations.
Binning-based approaches attempt to overcome
geometric rigidity by dividing the simulation cell into
a regular 3D grid and aggregating atomic properties—
Weakly Supervised Graph Neural Networks for Scalable 3D Phase Segmentation in Molecular Dynamics Simulations
303

typically counts or densities—within each bin. This
enables bulk statistical analysis of local composition
and has been used to infer regions with varying ele-
mental concentration. For example, Fe atom counts
across bins can yield bimodal distributions, where the
low and high count peaks correspond to Si-rich and
Fe-rich regions, respectively (Shakya et al., 2024).
Bins between the peaks are heuristically interpreted
as boundary regions. While this method provides a
coarser but interpretable classification of the domain,
it still lacks atomic-level granularity. Moreover, the
quality of the segmentation depends strongly on the
bin size and user-defined thresholds. These thresh-
olds do not necessarily reflect physical principles and
must be retuned for different system sizes or simula-
tion setups. The intermediate bins, although intended
to represent interfaces, are not explicitly modeled, and
the lack of learning mechanisms prevents generaliza-
tion or reuse across different datasets.
Machine learning-based techniques offer the po-
tential to overcome many of these limitations by
learning complex spatial and chemical patterns di-
rectly from data. Voxel-based 3D convolutional
neural networks (3D-CNNs), which operate on dis-
cretized atomic grids, have demonstrated success
in binary phase classification tasks such as solid-
liquid separation (Fukuya and Shibuta, 2020). How-
ever, convolutional filters in 3D-CNNs are structure-
agnostic and do not inherently incorporate domain-
specific information such as element identity or chem-
ical coordination, which are critical for distinguishing
compositionally complex phases. Also, these models
typically require fixed-size inputs, making them ill-
suited to variable-sized simulation cells.
GNNs address these challenges by working di-
rectly on atomic point clouds and leveraging chem-
ical and spatial relationships through neighborhood
graphs. Our method builds on this approach by incor-
porating weak supervision from a physics-informed
unsupervised clustering pipeline applied to small sys-
tems. We use kernel density estimation to smooth
atomic distributions, followed by density-based clus-
tering and morphological operations, to generate
coarse per-atom labels for Fe-rich, Fe-poor, and
boundary regions. These labels, derived from phys-
ically informed unsupervised analysis (Jadrich et al.,
2018), guide the GNN to learn associations between
local atomic environments and phase identity. Once
trained, the GNN supports efficient inference through
radius-based neighborhood graphs, scales linearly
with the number of atoms, and generalizes to much
larger systems with varying cell sizes and configu-
rations. Crucially, we model transition regions ex-
plicitly using a post-processing dilation step based on
average atomic bond lengths, yielding a more phys-
ically interpretable classification of boundary atoms.
This enables us to bridge the gap between discrete,
coarse-grained binning and fine-grained, learned per-
atom segmentation.
By combining weakly supervised labels, domain-
informed graph features, and scalable inference,
our method overcomes the limitations of geometric
heuristics, binning methods, CNN-based models, and
structure-based classifiers. It enables detailed per-
atom phase classification in chemically diverse, irreg-
ular systems—capturing boundaries with physical fi-
delity, eliminating manual threshold tuning, and scal-
ing efficiently to large simulations.
3 METHODOLOGY
Accurate phase segmentation in molecular dynam-
ics simulations requires addressing two fundamental
challenges: (1) the absence of ground truth labels for
training supervised models, and (2) the computational
intractability of applying accurate unsupervised meth-
ods to large systems. We propose a physics-guided
weak supervision framework that distills knowledge
from small-scale unsupervised analyses into a scal-
able GNNs . Our approach is motivated by three key
observations:
• Physical Priors Enable Weak Labeling. Al-
though manual labeling is impractical, the funda-
mental physics of phase separation imposes natu-
ral constraints that guide label generation:
– Fe-rich metal region and Fe-poor silicate re-
gions exhibit distinct density distributions.
– Interfacial widths are typically on the order of
atomic bond lengths.
These properties allow for automated label gen-
eration via density-based clustering and post-
processing.
• The Scalability Barrier. High-resolution unsu-
pervised methods such as kernel density estima-
tion scale as O(N · M), where N is the number of
atoms and M the number of voxels. As detailed
in Table 3, this scaling becomes a computational
bottleneck for large systems, limiting the feasibil-
ity of such methods for large-scale simulations.
• Local Environments Transfer Across Scales.
Atomic-level phase features—such as coordina-
tion numbers and local elemental ratios—are size-
invariant. This enables GNNs trained on weakly
labeled small systems to generalize to larger sys-
tems through learned local feature representa-
tions.
KDIR 2025 - 17th International Conference on Knowledge Discovery and Information Retrieval
304

Our solution combines these insights in a two-stage
process as shown in Figure 1.
3.1 Weak Label Generation Pipeline
We created a specialized weak labeling system to train
supervised learning models without requiring hand-
labeled training data, using principles from metal-
silicate phase separation physics. The objective is
to distinguish between Fe-rich (metallic) and Fe-
poor (silicate) areas within atomic-scale simulations.
Since clustering atoms based on their 3D positions
proves unreliable—due to the complex, non-linear
boundaries between phases—we converted the anal-
ysis from spatial coordinates to density-based rep-
resentations. This transformation makes the phase
boundaries more linearly defined, which significantly
improves the performance of unsupervised cluster-
ing algorithms. The pipeline includes these essential
components:
3.1.1 Density Field Construction
We compute a 3D voxelized Fe density field us-
ing kernel density estimation (KDE) (Silverman,
1986) over the atomic coordinates, as implemented
in SciPy (Virtanen et al., 2020). This transforms the
sparse atomic distribution into a smooth scalar field
that captures local Fe concentration, making spatial
patterns more discernible and suitable for clustering.
To properly account for the periodic nature of the sim-
ulation cell, we replicate Fe atom positions across ad-
jacent periodic images before applying KDE. This en-
sures that density is smoothly estimated near the sim-
ulation box boundaries, avoiding artificial discontinu-
ities. The simulation cell is discretized into a uniform
grid of 50 × 50 × 50 voxels, providing a balance be-
tween spatial resolution and computational cost. KDE
scales as O(N · M), where N is the number of atoms
and M the number of voxels. Hence, increasing voxel
resolution leads to a significant rise in computational
complexity. Our selected grid resolution captures the
relevant physical features while keeping density esti-
mation computationally tractable. Since this process
is repeated across thousands of simulation snapshots,
the total cost adds up substantially.
3.1.2 K-Means Clustering in Density Space
We apply K-Means clustering, specifying the num-
ber of clusters as two, to the voxelized density data,
partitioning the system into Fe-rich and Fe-poor do-
mains. Each voxel, which captures local iron con-
centration, serves as a data point in the scalar density
field. The clustering algorithm processes only vox-
els containing non-zero density values, and we des-
ignate the cluster exhibiting higher mean density as
the Fe-rich phase. This strategy exploits the inherent
bimodal density distribution characteristic of phase-
separated systems, where concentrated metallic re-
gions are distinctly separated from dispersed silicate
areas. The density-based approach transforms intri-
cate spatial boundaries into more manageable linear
separations, allowing a straightforward unsupervised
clustering method to achieve reliable phase identifica-
tion.
3.1.3 Periodic Boundary Condition-Aware
Connected Component Analysis
Clustering based solely on voxel density can produce
fragmented regions scattered throughout the simula-
tion cell. To enforce spatial coherence, we apply
connected component labeling to the Fe-rich clus-
ter assignments. This is implemented using a cus-
tom union-find algorithm that explicitly handles peri-
odic boundary conditions—a crucial consideration for
atomistic systems where atoms near the boundaries
may interact across simulation cell edges. Among all
Fe-rich regions identified, we retain only the largest
spatially connected component and designate it as
the metal phase. All remaining regions, including
smaller disconnected Fe-rich fragments, are classified
as part of the silicate phase, regardless of their local
iron concentration. This post-processing step aligns
with the physical expectation that the system consists
of exactly two macroscopic phases—metal and sili-
cate—separated by a single continuous interface. By
preserving only the dominant connected Fe-rich re-
gion, we ensure the metal phase is correctly captured
while avoiding spurious classification of isolated Fe-
rich pockets as separate metallic domains.
3.1.4 Boundary Region Assignment
To model the fuzzy transition zone between metal
and silicate phases, we apply binary dilation to both
the metal and silicate regions. Binary dilation is
a morphological operation that expands a region by
including neighboring voxels within a specified ra-
dius, effectively growing the mask outward and cap-
turing nearby space (Serra, 1982). When applied to
both regions independently, the overlapping volume
of their dilated masks defines the boundary region.
This boundary represents the interfacial zone where
atoms are likely influenced by both phases. While the
dilation radius is initially guided by typical atomic
bond lengths, the final boundary thickness is deter-
mined empirically. We evaluate the variance of Fe
weight percent in the Fe-rich region across multiple
Weakly Supervised Graph Neural Networks for Scalable 3D Phase Segmentation in Molecular Dynamics Simulations
305
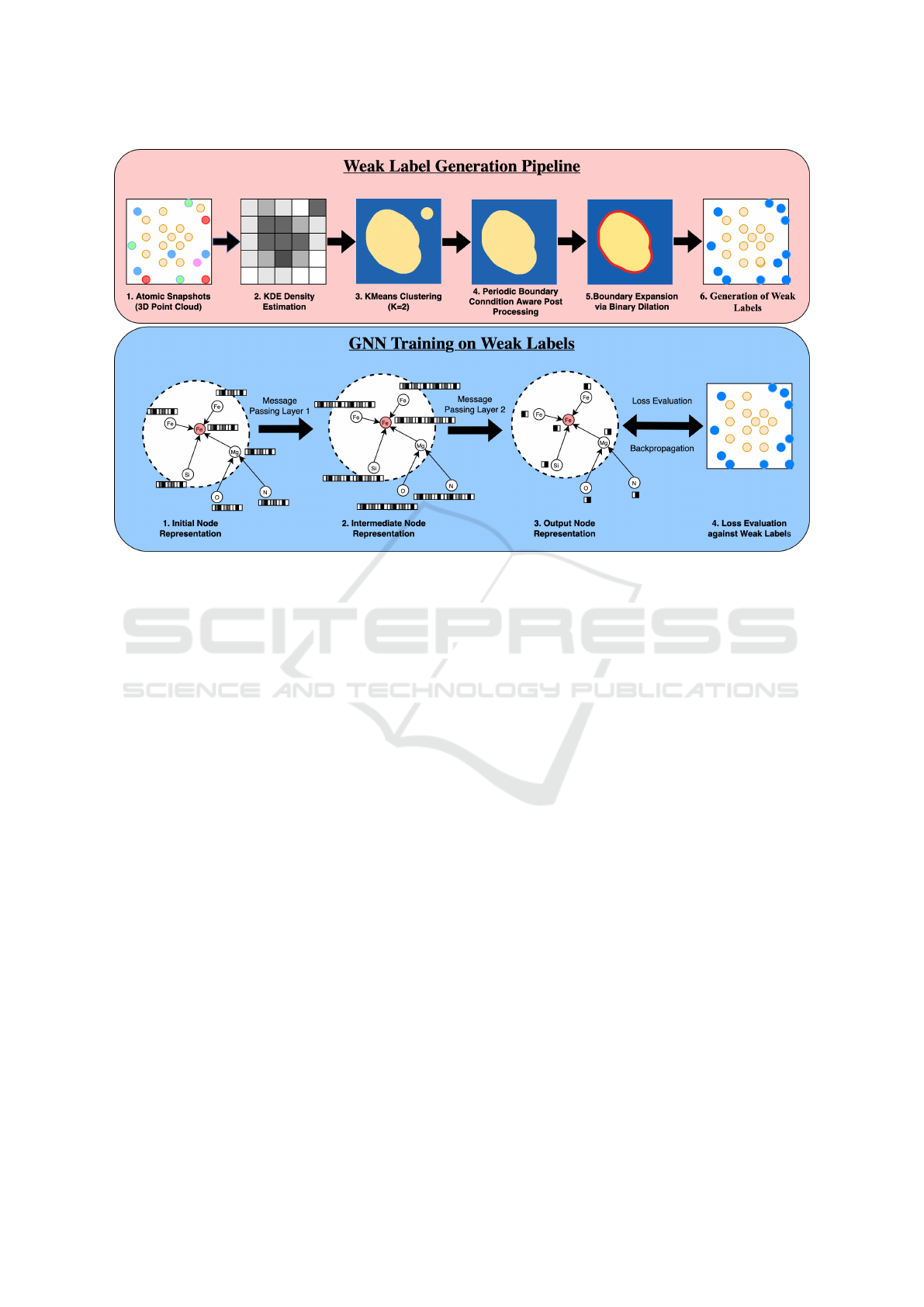
Figure 1: Schematic of the proposed phase segmentation approach. Top: Weak labels are generated via a pipeline that involves
KDE-based density estimation, clustering, Periodic Boundary Condition aware post-processing, and boundary (interface)
expansion using binary dilation. Bottom: A GNN is trained on these weak labels using node features based on atomic type
and local environment.
simulation snapshots for different boundary widths
and select the value that minimizes this variance, in-
dicating improved stability in phase classification and
reduced ambiguity at the interface.
3.1.5 Label Assignment to Atoms
To generate per-atom labels, we map each atomic
position to its corresponding voxel in the clustered
density grid. The voxel’s pre-assigned phase la-
bel—either Fe-rich metal, Fe-poor silicate, or bound-
ary—is transferred to all atoms whose positions fall
within that voxel. This mapping ensures consistency
between the density-based voxel segmentation and
the atom-level labels required for model training. Be-
cause the voxel grid spans the entire simulation cell,
every atom is assigned a label based on its spatial lo-
cation relative to the phase-separated structure. This
process results in a fully labeled dataset of atoms,
where each atom inherits the phase identity of its local
environment as inferred from the KDE-based cluster-
ing and post-processing pipeline. These labels serve
as weak supervision targets for training our graph
neural network model.
3.2 GNN Training on Weak Labels
We formulate a semantic phase segmentation as a
node classification task on atomic graphs, where su-
pervised learning is performed using weak labels de-
rived from our physics-informed pipeline. This ap-
proach tests whether a lightweight graph neural net-
work can replicate the accuracy of our computation-
ally intensive density-based method while achieving
superior scalability. To evaluate the generality of
our learning framework, we implement two different
GNN architectures—GCN and GAT—using identical
feature inputs and training protocols. This compara-
tive setup allows us to assess whether our weak super-
vision strategy is compatible with different message-
passing schemes.
3.2.1 Graph Representation and Node Features
Each atomic configuration is represented as an undi-
rected graph G = (V, E), where each node v
i
∈ V cor-
responds to an atom, and an edge (i, j) ∈ E connects
atoms i and j if their spatial separation is less than a
predefined cutoff distance r
c
. To preserve the phys-
ical continuity of the atomic environment, we apply
periodic boundary conditions (PBC) during neighbor
search using a wrapped distance metric.
Each node v
i
is associated with a feature vector
KDIR 2025 - 17th International Conference on Knowledge Discovery and Information Retrieval
306

x
i
∈ R
10
that encodes both chemical identity and local
structural information. The full node feature vector is
expressed as:
x
i
= concat
OneHot(type
i
), N
i
Fe
, µ
i
Fe
, σ
2
Fe
, f
i
Fe
, N
i
Mg
,
(1)
where N
i
Fe
denotes the number of Fe neighbors, µ
i
Fe
and σ
2
Fe
are the mean and variance of their distances,
f
i
Fe
is the fraction of Fe neighbors relative to total
neighbors, and N
i
Mg
is the count of Mg neighbors.
These features are chosen to reflect both the chem-
ical identity of the central atom and the local coordi-
nation environment, which are known to be predictive
of phase identity in metal-silicate systems. In par-
ticular, statistics over Fe neighbors help capture the
density and spatial distribution of metallic bonding,
while the inclusion of Mg neighbors aids in identify-
ing silicate-like environments. Neighbor counts for
Si and O were initially considered but later excluded
from the final feature design, as they were found to be
redundant. Their inclusion had negligible impact on
the macro F1 score for GCN (0.878 vs. 0.873 without
them) and slightly reduced the performance of GAT
(0.809 vs. 0.819 without them), indicating that these
features do not contribute meaningfully to the phase
classification task.
3.2.2 Message Passing and Prediction
Framework
GNNs operate by iteratively updating node represen-
tations through localized neighborhood aggregation.
For a given graph G = (V, E), with initial node fea-
tures h
(0)
i
= x
i
, each GNN layer refines node embed-
dings via:
h
(l+1)
i
= σ
AGG
(l)
n
h
(l)
j
| j ∈ N (i)
o
∪
n
h
(l)
i
o
,
(2)
where N (i) denotes the set of neighbors of node i,
σ is the ReLU activation function, and AGG
(l)
is the
layer-specific aggregation function.
Our model architecture consists of two such
message-passing layers, enabling nodes to incorpo-
rate information from both first and second-order
neighborhoods. The output of the final layer is a 2-
dimensional embedding used for binary classification
(Fe-rich vs. Fe-poor). These logits are converted into
class probabilities using a softmax function:
ˆ
y
i
= softmax(h
(L)
i
), (3)
where L is the total number of layers.
We implement and evaluate two GNN variants,
each employing a different aggregation strategy:
GraphConv: The GraphConv operator uses nor-
malized aggregation as follows:
h
(l+1)
i
= σ
∑
j∈N (i)∪{i}
1
p
|N (i)| |N ( j)|
W
(l)
h
(l)
j
.
(4)
GAT: Graph Attention Networks introduce learn-
able attention weights between neighbors:
h
(l+1)
i
= σ
∑
j∈N (i)
α
(l)
i j
W
(l)
h
(l)
j
, (5)
with attention coefficients computed as:
α
(l)
i j
=
exp
f
a
⊤
h
W
(l)
h
(l)
i
∥ W
(l)
h
(l)
j
i
∑
k∈N (i)
exp
f
a
⊤
h
W
(l)
h
(l)
i
∥ W
(l)
h
(l)
k
i
(6)
where W
(l)
∈ R
d
′
×d
is a learnable weight matrix,
a ∈ R
2d
′
is a learnable attention weight vector, ∥ de-
notes vector concatenation, and f is the LeakyReLU
activation function.
Training Objective. The network is trained using
the standard cross-entropy loss, defined over the set
of weakly labeled atoms:
L = −
∑
i∈T
2
∑
c=1
y
ic
log ˆy
ic
, (7)
where T is the set of labeled atoms, y
ic
∈ {0, 1} is
the one-hot encoded ground truth weak label, and ˆy
ic
is the predicted class probability from Equation 3.
Training is performed using the Adam optimizer, with
both models trained under the same hyperparameter
settings.
4 EXPERIMENTS AND RESULTS
4.1 Weak Label Generation
We first evaluate the effectiveness of our weak la-
beling pipeline on a small FeMgSiON system con-
taining 520 atoms in a cubic simulation supercell
of 17
˚
A length. Each atom is assigned a phase
label—Fe-rich (metal), Fe-poor (silicate), or bound-
ary/interface—based on its spatial location within a
voxelized grid, where each voxel is classified into one
of the three regions. Atoms inherit the phase label of
the voxel they fall into.
Weakly Supervised Graph Neural Networks for Scalable 3D Phase Segmentation in Molecular Dynamics Simulations
307
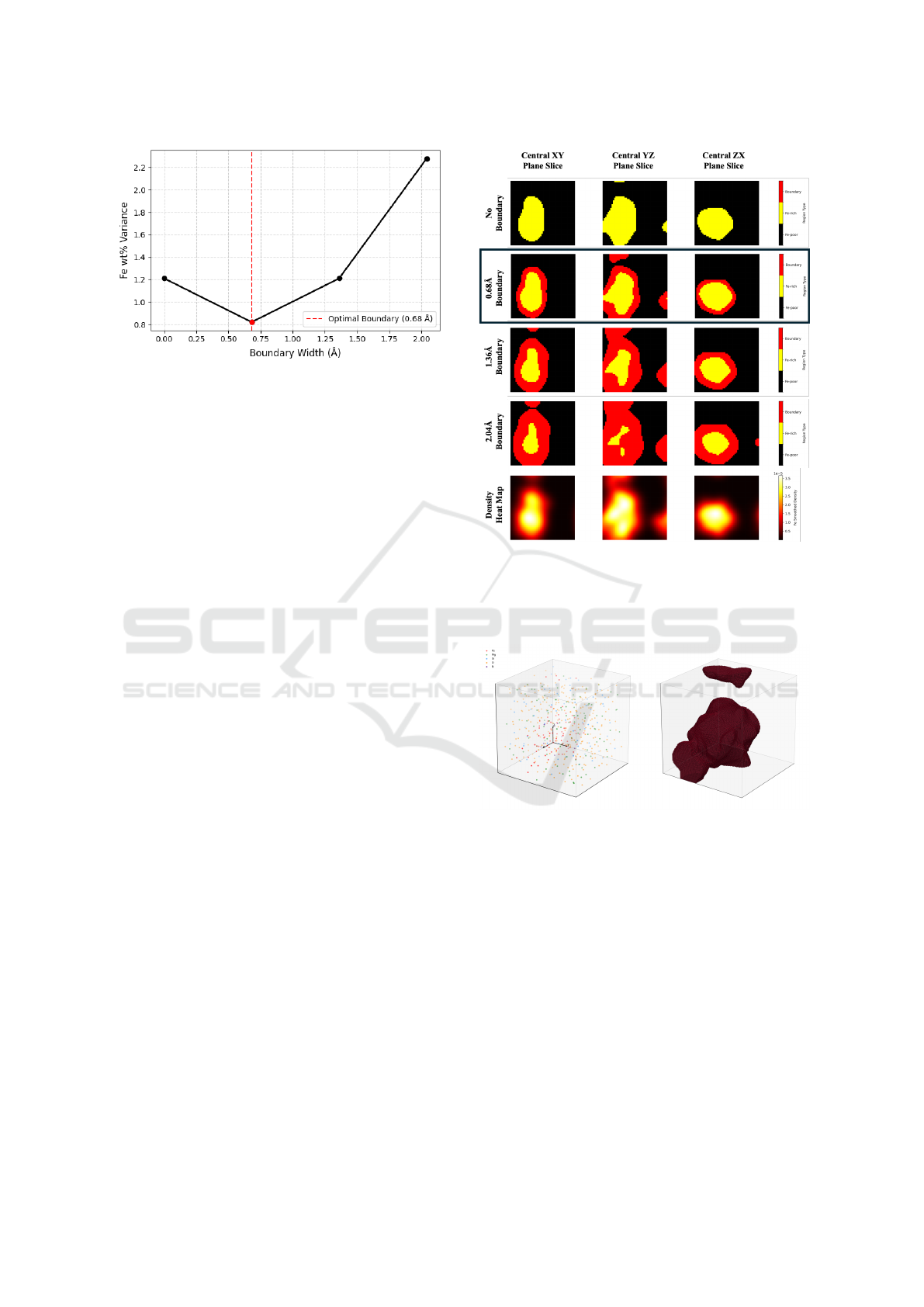
Figure 2: Variance of Fe weight percent in the Fe-rich re-
gion across 1000 snapshots from the 3000 K simulation,
plotted as a function of boundary width.
A critical hyperparameter in this labeling process
is the boundary width, which determines how many
voxels are designated as interfacial (boundary) rather
than purely Fe-rich or Fe-poor. To determine an ap-
propriate value, we evaluate the variance of Fe weight
percent in the Fe-rich region across 1000 simulation
snapshots, under different boundary thicknesses. As
shown in Figure 2, a boundary width of 0.68
˚
A re-
sults in the lowest variance, indicating greater stabil-
ity in the phase classification and reduced ambiguity
at the interface. While this analysis was conducted at
3000 K, we found that the same boundary width also
yielded the lowest variance for simulations at 4000 K.
We visualize how different boundary widths affect
segmentation outcomes in Figure 3, which shows cen-
tral XY, YZ, and XZ slices of a representative snap-
shot. Without a boundary region, the segmentation is
overly sharp and fails to capture the transitional na-
ture of the interface. Introducing a 0.68
˚
A boundary
produces smoother, more physically realistic segmen-
tation. Larger boundaries (1.36
˚
A, 2.04
˚
A) begin to
erode phase interiors, reducing fidelity.
To assess the spatial coherence and physical plau-
sibility of the assigned labels, we visualize the Fe-rich
region from a reference snapshot. As shown in Fig-
ure 4, the segmented metallic domain forms a large,
continuous structure consistent with physical expec-
tations for phase-separated systems. Although the vi-
sualization shows two disconnected volumes, they be-
long to a single contiguous phase, split only by peri-
odic boundaries. Our labeling method accounts for
this, correctly identifying such regions as topologi-
cally connected. This confirms that the chosen bound-
ary width and labeling approach preserve spatial con-
tinuity and yield stable, physically meaningful phase
assignments suitable for training downstream models.
Figure 3: Phase segmentation slices across three planes
(XY, YZ, XZ) for different boundary sizes in the 3000 K
simulation. Rows show no boundary, 0.68
˚
A, 1.36
˚
A, and
2.04
˚
A, respectively. The 0.68
˚
A boundary best preserves
phase boundaries without distorting region interiors.
Figure 4: 3D visualization of phase segmentation. Left:
Atom positions with species color-coded. Right: Seg-
mented Fe-rich region rendered as a connected volume. The
split appearance is due to periodic boundaries; the region is
physically continuous.
4.2 Phase Segmentation with
Graph-Based Models
We conducted experiments on two distinct FeMg-
SiON systems—one at 29 GPa and 3000 K, and an-
other at 35 GPa and 4000 K. For each condition, sim-
ulations were performed at two different scales. Each
system was simulated at two scales: a small system
(520 atoms) used for training, and a large system
(33,280 atoms) used for inference and validation. The
small and large systems were constructed with identi-
KDIR 2025 - 17th International Conference on Knowledge Discovery and Information Retrieval
308
cal elemental ratios, differing only in spatial scale and
total number of atoms.
For each temperature-pressure condition, we
trained GNNs independently on the small system.
Specifically, we used two architectures—GAT and
GCN—and evaluated their performance on the corre-
sponding large system. We benchmarked their out-
puts against labels generated by our unsupervised
pipeline, which serves as a high-fidelity—but com-
putationally expensive reference.
4.3 Results and Analysis
To evaluate classification performance, we tested two
GNN architectures—GAT and GCN—on large-scale
FeMgSiON systems at two thermodynamic states.
Both models were trained on small systems using
weak labels derived from our unsupervised segmen-
tation pipeline and evaluated on larger systems using
the following metrics:
• Elemental weight percent (wt%) in predicted Fe-
rich (metal) and Fe-poor (silicate) regions, aver-
aged over 100 snapshots,
• Classification accuracy and F1 scores (micro,
macro, and per-class),
• ROC curves and confusion matrices for model
comparison.
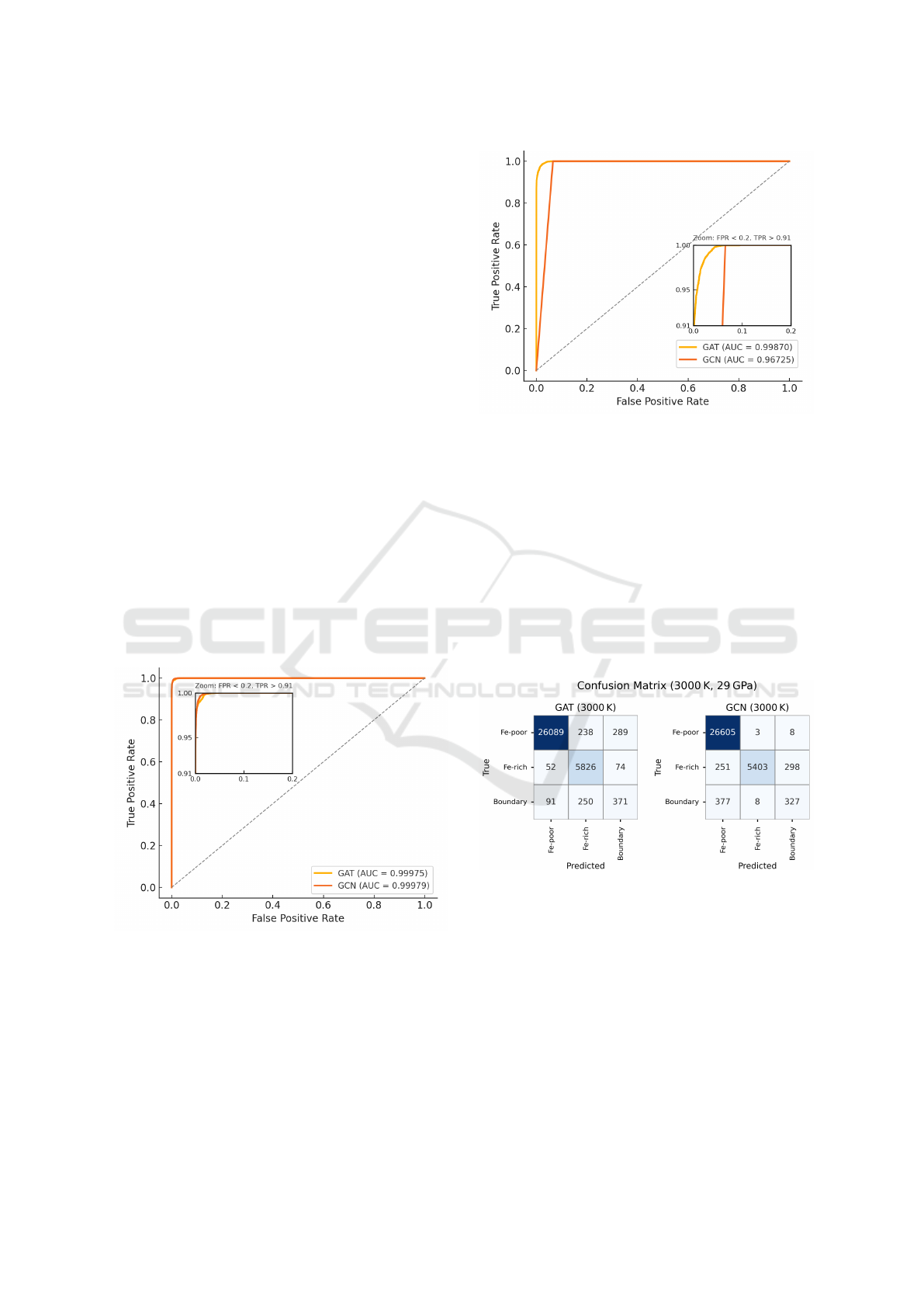
Figure 5: ROC curve comparing the classification perfor-
mance of GAT and GCN models on the reference snapshot
of the 3000 K, 29 GPa simulation. The inset highlights the
region with low false positive rates and high true positive
rates.
At 3000 K, where thermal agitation is minimal
and phase boundaries are sharply defined, both GAT
and GCN generalize well from the small training sys-
tem to the large-scale target. The confusion matri-
ces (Figure 7) show strong diagonal dominance, and
Figure 6: ROC curve comparing the classification perfor-
mance of GAT and GCN models on the reference snapshot
of the 4000 K, 35 GPa simulation. The inset highlights the
region with low false positive rates and high true positive
rates, showing finer differences in model sensitivity.
the ROC curves (Figure 5) confirm near-perfect sep-
arability, with area under the curve (AUC) exceeding
0.999 for both models. These high AUC values reflect
threshold-agnostic discriminative performance, indi-
cating that both GNNs reliably distinguish Fe-rich,
Fe-poor, and boundary atoms across a range of thresh-
olds. The macro F1 scores also exceed 0.85, under-
scoring consistent performance across all classes (Ta-
ble 1).
Figure 7: Confusion matrices for Fe-rich, Fe-poor, and
boundary classification at 3000 K and 29 GPa using GAT
and GCN. Shown for the reference snapshot of our large-
scale simulation (33,280 atoms), predictions are bench-
marked against labels obtained from the high-fidelity un-
supervised segmentation pipeline.
At 4000 K, increased thermal mixing introduces
ambiguity in phase boundaries, making segmenta-
tion more difficult. GCN’s performance degrades sig-
nificantly, particularly in its ability to detect bound-
ary regions, as reflected in a low boundary F1 score
(0.06). Its AUC also drops to 0.967, indicating re-
duced confidence in its ranking. While GCN cor-
rectly classifies nearly all Fe-rich (Metal) phase atoms
Weakly Supervised Graph Neural Networks for Scalable 3D Phase Segmentation in Molecular Dynamics Simulations
309
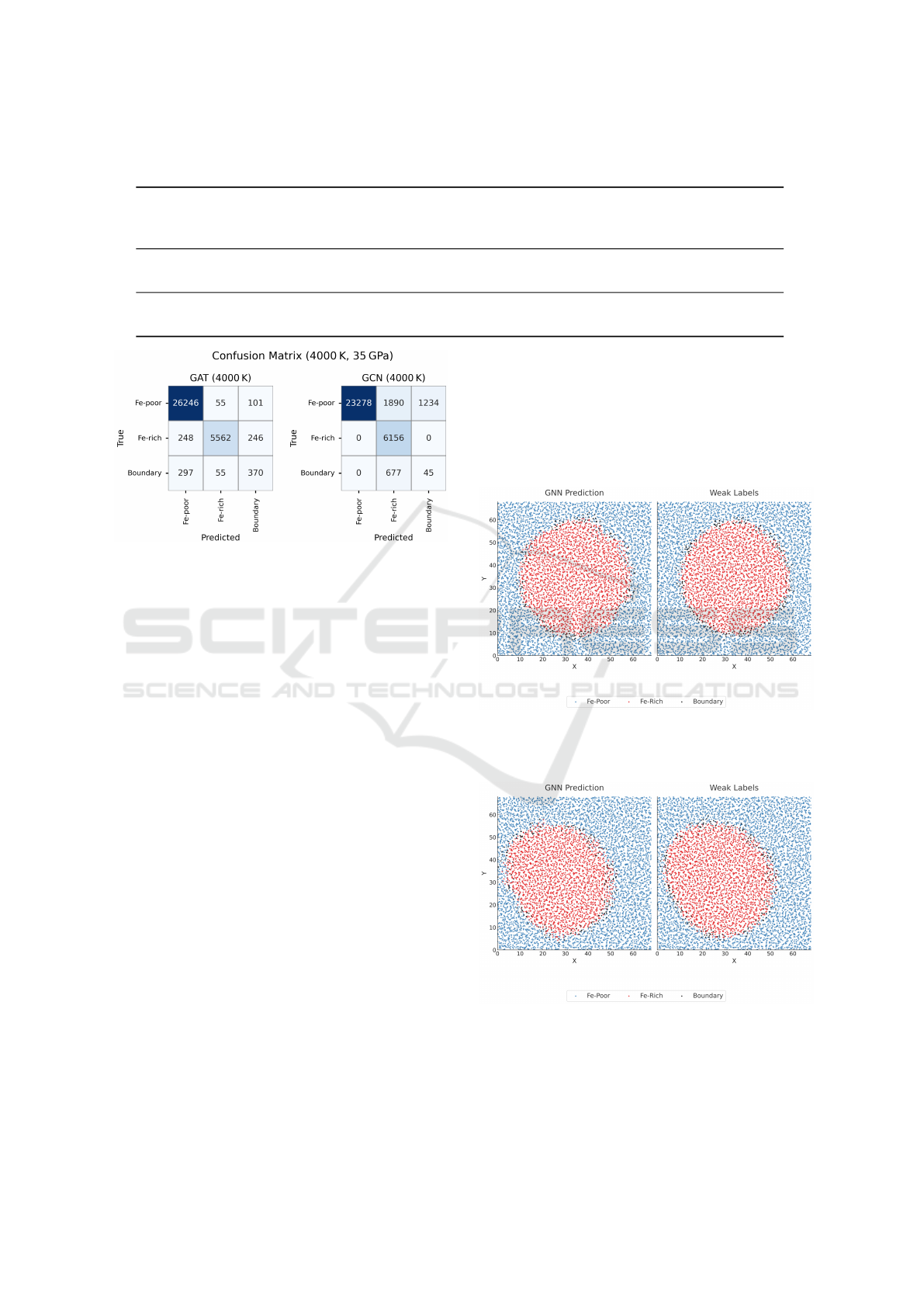
Table 1: Classification performance of GNN models across systems. Reported values include accuracy, class-wise F1 scores,
macro F1, and micro F1 scores.
System Model Accuracy
F1
(Fe-Poor/
Silicate)
F1
(Fe-Rich/
Metal)
F1
(Boundary)
Macro
F1
Micro
F1
3000 K, 29.1 GPa
GAT 0.979 0.986 0.964 0.506 0.819 0.979
GCN 0.986 0.995 0.945 0.678 0.873 0.986
4000 K, 35 GPa
GAT 0.975 0.986 0.929 0.515 0.810 0.975
GCN 0.899 0.926 0.866 0.061 0.618 0.899
Figure 8: Confusion matrices for Fe-rich, Fe-poor, and
boundary classification at 4000 K and 35 GPa using GAT
and GCN. Shown for the reference snapshot of our large-
scale simulation (33,280 atoms), predictions are bench-
marked against labels obtained from the high-fidelity un-
supervised segmentation pipeline.
at 4000 K (see Figure 8), it fails to resolve transi-
tional regions, misclassifying most boundary atoms
as Fe-rich—highlighting its limited capacity to cap-
ture compositional shifts in disordered systems. In
contrast, GAT maintains strong performance, with
an AUC above 0.998 and a macro F1 of 0.81. We
attribute this robustness to its attention mechanism,
which adaptively weighs contributions from neigh-
boring atoms and remains effective even under high-
temperature, disordered conditions.
Table 2 summarizes the average elemental com-
positions (wt%) predicted for Fe-rich/Metal and Fe-
poor/Silicate phases under both 3000 K/29 GPa and
4000 K/35 GPa conditions. At 3000 K, both GNN
models closely match the unsupervised baseline, in-
dicating accurate phase segmentation under low mix-
ing. At 4000 K, GCN predictions deviate more no-
ticeably from the unsupervised results—particularly
in Fe and Mg content—while GAT continues to pro-
duce more consistent estimates. These trends high-
light the improved reliability of GAT in capturing
phase behavior under more challenging thermody-
namic conditions.
Figures 9 and 10 show the XY projections of
atoms whose Z-bins lie between 20 and 30 from the
reference test snapshots of the large-scale systems at
29 GPa and 3000 K, and 35 GPa and 4000 K, respec-
tively. In both cases, this representative slice is ex-
tracted from a 50×50×50 spatial grid. These visual-
izations compare the GNN predictions against weak
labels to qualitatively assess the model’s ability to re-
cover physically meaningful phase separation into Fe-
rich, Fe-poor, and boundary regions.
Figure 9: XY projection of atoms from the central Z slice
of our reference snapshot in the 3000 K, 29 GPa system,
comparing GNN predictions with weak labels.
Figure 10: XY projection of atoms from the central Z slice
of our reference snapshot in the 4000 K, 35 GPa system,
comparing GNN predictions with weak labels.
Beyond segmentation accuracy, a key advantage
of our graph-based approach is its speed at inference
time. In particular, we compare the inference time of
KDIR 2025 - 17th International Conference on Knowledge Discovery and Information Retrieval
310

Table 2: Average elemental composition (wt%) in Fe-rich (Metal) and Fe-poor (Silicate) regions for both sys-
tems—3000 K/29 GPa and 4000 K/35 GPa—as predicted by GAT, GCN, and the unsupervised baseline. Values are averaged
over 100 test snapshots; standard error is reported.
T–P Phase Method Fe (wt %) Mg (wt %) Si (wt %) O (wt %) N (wt %)
3000 K, 29 GPa
Fe-rich
(Metal)
GAT 93.22 ± 0.18 0.06 ± 0.18 1.61 ± 0.10 1.83 ± 0.07 3.20 ± 0.00
GCN 94.64 ± 0.20 0.00 ± 0.00 1.57 ± 0.12 0.72 ± 0.08 3.05 ± 0.02
Unsupervised 93.81 ± 0.21 0.10 ± 0.01 1.67 ± 0.12 1.27 ± 0.09 3.15 ± 0.01
Fe-poor
(Silicate)
GAT 1.23 ± 0.07 29.45 ± 0.02 23.06 ± 0.05 46.13 ± 0.03 0.11 ± 0.00
GCN 5.06 ± 0.12 28.04 ± 0.02 22.06 ± 0.06 44.56 ± 0.05 0.28 ± 0.00
Unsupervised 3.77 ± 0.08 28.53 ± 0.02 22.39 ± 0.05 45.10 ± 0.03 0.19 ± 0.00
4000 K, 35 GPa
Fe-rich
(Metal)
GAT 90.59 ± 0.03 0.10 ± 0.01 2.99 ± 0.03 3.06 ± 0.04 3.25 ± 0.01
GCN 80.27 ± 0.25 3.41 ± 0.10 5.11 ± 0.05 8.41 ± 0.14 2.87 ± 0.01
Unsupervised 89.54 ± 0.01 0.37 ± 0.01 3.35 ± 0.03 3.52 ± 0.02 3.22 ± 0.02
Fe-poor
(Silicate)
GAT 3.47 ± 0.04 29.28 ± 0.01 22.07 ± 0.01 45.03 ± 0.02 0.14 ± 0.01
GCN 2.17 ± 0.04 29.82 ± 0.04 22.30 ± 0.06 45.68 ± 0.06 0.08 ± 0.00
Unsupervised 5.79 ± 0.05 28.49 ± 0.02 21.47 ± 0.02 44.07 ± 0.01 0.19 ± 0.01
our trained GNNs against the original unsupervised
pipeline used for generating weak labels. All infer-
ences were conducted on the 33,280-atom large-scale
system using non-parallelized implementations. To
ensure fair comparison, all methods were executed on
identical hardware with the same processor configu-
ration.
Table 3: Inference time and scaling behavior of different
segmentation methods, reported over 100 test snapshots
from the 33,280-atom large-scale system. Values reflect the
mean and standard error.
Method Time (s) Scaling
GNN (GAT) 3.61 ± 0.12 ∼ O(N)
GNN (GCN) 3.12 ± 0.14 ∼ O(N)
Unsupervised 210.0 ± 2.2 O(N · M)
(KDE + Clustering)
GNN inference comprises two steps: graph con-
struction and forward propagation. Graph construc-
tion uses a fixed-radius neighbor search with spatial
indexing (e.g., cKDTree), scaling approximately lin-
early with system size. The forward pass involves a
constant number of message-passing layers and also
scales linearly with the number of atoms. In contrast,
the unsupervised pipeline involves kernel density es-
timation (KDE) over a 3D voxel grid, followed by
clustering and morphological post-processing. KDE
requires each atom to contribute to many voxels, re-
sulting in O(N · M) complexity, where N is the num-
ber of atoms and M the number of voxels—making
it a computational bottleneck for large-scale simula-
tions.
As shown in Table 3, our GNN models reduce
inference time from over 200 seconds to just over
3 seconds for the 33,280-atom system—achieving a
speedup of over two orders of magnitude. This high-
lights their efficiency over the original unsupervised
pipeline and suitability for large-scale applications.
5 CONCLUSIONS
We introduced a hybrid learning approach for scal-
able and accurate phase segmentation in large-scale
molecular dynamics simulations by combining a
structure-aware unsupervised pipeline with a weakly-
supervised GNNs. This approach enables model
training even in the absence of labeled data by lever-
aging structural heuristics to generate weak supervi-
sion. The GNNs are trained on small systems but gen-
eralize effectively to much larger configurations with-
out sacrificing accuracy, demonstrating robust perfor-
mance across varying system sizes. Among the ar-
chitectures evaluated, GATs in particular showed con-
sistent performance across systems with different de-
grees of disorder, effectively capturing boundary re-
gions. As parametric models, they offer significant
speedups during inference by eliminating the need
for repeated unsupervised computations, with runtime
benefits that grow with system size. While demon-
strated on FeMgSiON systems, the strategy is broadly
applicable to other multi-phase materials where high-
quality labels are unavailable but structural cues ex-
ist. These results underscore a broader opportunity
in using machine learning to accelerate and scale sci-
entific analyses in domains where conventional la-
beling is impractical. A promising direction for
future work is the integration of uncertainty-aware
active learning, where the model identifies regions
of low confidence—particularly near phase bound-
aries—and selectively queries for additional weak su-
pervision. Techniques such as Monte Carlo Dropout
Weakly Supervised Graph Neural Networks for Scalable 3D Phase Segmentation in Molecular Dynamics Simulations
311

or Bayesian GNNs could be employed to estimate
uncertainty, allowing the model to prioritize ambigu-
ous regions and further improve segmentation quality
while minimizing labeling overhead.
ACKNOWLEDGEMENTS
This work was supported by NASA (Grant No.
80NSSC21K0377) and the National Science Founda-
tion (EAR 1463807). Computational resources were
provided by the High Performance Computing facil-
ity at Louisiana State University. Additional support
was received through the Summer Opportunities Fel-
lowship, awarded by Shell Oil Company.
REFERENCES
Chazelle, B. (1993). An optimal convex hull algorithm in
any fixed dimension. Discrete & Computational Ge-
ometry, 10(4):377–409.
Chen, L.-C., Papandreou, G., Kokkinos, I., Murphy, K.,
and Yuille, A. L. (2017). Rethinking atrous convo-
lution for semantic image segmentation. IEEE Trans-
actions on Pattern Analysis and Machine Intelligence
(TPAMI), 39(12):2341–2355.
Edelsbrunner, H. and M
¨
ucke, E. P. (1994). Three-
dimensional alpha shapes. ACM Transactions on
Graphics (TOG), 13(1):43–72.
Fukuya, T. and Shibuta, Y. (2020). Machine learning ap-
proach to automated analysis of atomic configuration
of molecular dynamics simulation. Computational
Materials Science, 184:109880.
Hamilton, W. L., Ying, R., and Leskovec, J. (2017). In-
ductive representation learning on large graphs. In
Advances in Neural Information Processing Systems
(NeurIPS).
Honeycutt, J. and Andersen, H. (1987). Molecular dynam-
ics study of melting and freezing of small lennard-
jones clusters. The Journal of Physical Chemistry,
91(19):4950–4963.
Jadrich, R. B., Lindquist, B. A., and Truskett, T. M. (2018).
Unsupervised machine learning for detection of phase
transitions. Physical Review E, 97:023301.
Kipf, T. N. and Welling, M. (2017). Semi-supervised clas-
sification with graph convolutional networks. In In-
ternational Conference on Learning Representations
(ICLR).
Long, J., Shelhamer, E., and Darrell, T. (2015). Fully con-
volutional networks for semantic segmentation. In
Proceedings of the IEEE Conference on Computer Vi-
sion and Pattern Recognition (CVPR), pages 3431–
3440.
Lopez, C. A., Vesselinov, V. V., Gnanakaran, S., and
Alexandrov, B. S. (2019). Unsupervised machine
learning for analysis of coexisting lipid phases and
domain growth in biological membranes. bioRxiv,
527630(v2). Preprint, version 2.
MacQueen, J. (1967). Some methods for classification and
analysis of multivariate observations. In Proceed-
ings of the Fifth Berkeley Symposium on Mathematical
Statistics and Probability, Volume 1: Statistics, pages
281–297. University of California Press.
McDonough, W. F. and Sun, S.-S. (1995). The composition
of the Earth. Chemical Geology, 120(3–4):223–253.
Qi, C. R., Su, H., Mo, K., and Guibas, L. J. (2017). Pointnet:
Deep learning on point sets for 3d classification and
segmentation. In Proceedings of the IEEE Conference
on Computer Vision and Pattern Recognition (CVPR),
pages 652–660.
Ronneberger, O., Fischer, P., and Brox, T. (2015). U-net:
Convolutional networks for biomedical image seg-
mentation. Medical Image Computing and Computer-
Assisted Intervention (MICCAI), 9351:234–241.
Scarselli, F., Gori, M., Tsoi, A. C., Hagenbuchner, M.,
and Monfardini, G. (2009). The graph neural net-
work model. IEEE Transactions on Neural Networks,
20(1):61–80.
Serra, J. (1982). Image analysis and mathematical morphol-
ogy. Academic Press.
Shakya, A., Ghosh, D. B., Jackson, C., Morra, G., and
Karki, B. B. (2024). Insights into core–mantle differ-
entiation from bulk earth melt simulations. Scientific
Reports, 14(1):18739.
Silverman, B. W. (1986). Density Estimation for Statistics
and Data Analysis. Chapman and Hall/CRC.
Stukowski, A. (2012). Structure identification methods for
atomistic simulations of crystalline materials. Mod-
elling and Simulation in Materials Science and Engi-
neering, 20(4):045021.
Veli
ˇ
ckovi
´
c, P., Cucurull, G., Casanova, A., Romero, A., Li
`
o,
P., and Bengio, Y. (2018). Graph attention networks.
In International Conference on Learning Representa-
tions (ICLR).
Virtanen, P., Gommers, R., Oliphant, T. E., and ... (2020).
SciPy 1.0: Fundamental algorithms for scientific com-
puting in python. Nature Methods, 17:261–272.
Zhang, Y. and Guo, G. (2009). Partitioning of si and o
between liquid iron and silicate melt: A two-phase
ab initio molecular dynamics study. Geophysical Re-
search Letters, 36(18):L18305.
KDIR 2025 - 17th International Conference on Knowledge Discovery and Information Retrieval
312
