
Multiple Sequence Alignment Using Ant Colony Optimization with
Chaotic Jump
Matheus Lino de Freitas
a
, Matheus Carreira Andrade
b
, Anderson Rici Amorim
c
,
Vitoria Zanon Gomes
d
, Bruno Rodrigues da Silveira
e
, Gabriel Augusto Prevato
f
,
Luiza Guimar
˜
aes Cavarc¸an
g
, Carlos Roberto Val
ˆ
encio
h
and Geraldo Francisco Doneg
´
a Zafalon
i
Department of Computer Science and Statistics, Universidade Estadual Paulista (UNESP),
Rua Crist
´
ov
˜
ao Colombo, 2265, Jardim Nazareth, S
˜
ao Jos
´
e do Rio Preto - SP, 15054-000, Brazil
Keywords:
Multiple Sequence Alignment, Ant Colony Optimization, Hybrid Approaches, Chaotic Jump.
Abstract:
Multiple sequence alignment is one of the most relevant techniques in the bioinformatics. The next generation
sequencing technologies produces a large volume of data that is later analised by biologists, biomedicals and
geneticists. Due to this huge volume, computational effort are necessary to aid in the data analisys, as an
example, for sequence alignment. This works aims to introduce a novel method that combines the KAlign and
Clustal Omega tools in order to produce a seed alignment that will later be refined by Ant Colony Optimization
and Chaotic Jump. The results showed that for every test the ACO produced better alignments than the MSA-
GA tool and at least for 50% of tests the proposed method was able to improve the initial alignments produced
by the KAlign and Clustal Omega tools.
1 INTRODUCTION
After the discovery of the chemical structure of the
deoxyribonucleic acid (DNA) molecule in 1953, the
genetic code and the flow of biological information
from the molecules, a new area, bioinformatics, oc-
curred. This area involves several lines of knowledge
and has the fundamental role of providing computa-
tional solutions to various real biological problems
(Prosdocimi et al., 2002). After more than 30 years
this area has gained more and more expressiveness,
thanks to the evolution of hardwares and the growth
in the number of information available, but there are
still many problems to be faced (Zafalon, 2009). Con-
sequently, sequence alignments have become relevant
and necessary for the analysis of this large amount of
a
https://orcid.org/0009-0004-7969-5729
b
https://orcid.org/0000-0002-1670-266X
c
https://orcid.org/0000-0001-7862-7530
d
https://orcid.org/0000-0003-4176-566X
e
https://orcid.org/0009-0003-5941-9869
f
https://orcid.org/0009-0001-7091-9763
g
https://orcid.org/0009-0007-9589-3217
h
https://orcid.org/0000-0002-9325-3159
i
https://orcid.org/0000-0003-2384-011X
information (Sievers and Higgins, 2014). With this,
the present work proposes a contribution to a concept
widely used in bioinformatics, which is the multiple
alignment of sequences.
Because sequence alignment is from the prob-
lem class called Non-Deterministic Polynomial-Time
Hard or NP-Hard, the exact and even approximate
approaches are not used for large volumes of se-
quences (Zafalon et al., 2021). They use heuristics
and stochastic models, because these approaches can
find solutions with good biological quality within a
feasible time. Even heuristics may not converge to
a result considered acceptable because they can stag-
nate at local maximum or minimum points. There-
fore, in addition to a good model that helps prevent
these situations, chaotic approaches, for example, are
also applied for this purpose in the literature.
In addition, hybrid solutions are another option
used to improve the expected result. The work of
Lee et al. (2008) proposes a hybrid approach using
Genetic Algorithm for construction of alignment and
optimization by ant colony for refinement. Another
line of hybrid solution is the combination of tools,
as can be seen in the Rubio-Largo et al. (2016) work
that uses the KAlign tool to perform a realignment
of sequence fragments. The present work takes these
Lino de Freitas, M., Andrade, M. C., Amorim, A. R., Gomes, V. Z., Rodrigues da Silveira, B., Prevato, G. A., Cavarçan, L. G., Valêncio, C. R. and Zafalon, G. F. D.
Multiple Sequence Alignment Using Ant Colony Optimization with Chaotic Jump.
DOI: 10.5220/0013293800003929
In Proceedings of the 27th International Conference on Enterprise Information Systems (ICEIS 2025) - Volume 2, pages 237-244
ISBN: 978-989-758-749-8; ISSN: 2184-4992
Copyright © 2025 by Paper published under CC license (CC BY-NC-ND 4.0)
237

practical questions into consideration and proposes
a solution for multiple alignment of sequences us-
ing a hybrid solution with two tools: KAling (Lass-
mann, 2019) and Clustal Omega (Sievers and Hig-
gins, 2014) together with an optimization refinement
by ant colony combined with chaotic jump in order
to reduce the probability of reaching local maximums
and achieve higher quality results.
The aim of this work is to present a method for
multiple alignment of sequences, using the meta-
heuristic Ant Colony Optimization and seeking to ob-
tain results that are biologically relevant. Thus, hybrid
strategies were employed, such as the combination
of tools for generating a base alignment, refinement
of the alignment through the meta-heuristic cited and
application of the concept of chaotic jump combined
with partial realignment of sequences, if the algorithm
enters a local maximum point.
This work is organized as follows: In Section 2,
we present the related works, In Section 3 we detail
our methodology to develop the approach. In Section
4, we show the results of our method, focusing on
time execution improvement. Finally, in Section 5,
we make our conclusions about the work.
2 RELATED WORK
2.1 Multiple Sequence Alignment Based
on Chaotic PSO
The work of Lei et al. (2009) presents a multi-
sequence alignment approach, combining chaotic
jump with Particle Swarm Optimization. The authors
propose this solution to deal with the problem of pre-
mature convergence, which often means that the al-
gorithm has reached a local maximum or minimum
point (Gomes et al., 2022). Basically, the proposed
method perceives premature convergence by observ-
ing two points, first if the average distance of particles
is less than a threshold and second if the variance of
the particle satisfaction function is less than another
parameter of threshold.
When identifying the situation of premature con-
vergence, the algorithm uses a logistic map function
to generate values in the chaotic interval between 0
and 1. These values are then mapped to entire posi-
tions corresponding to gaps that will be inserted into
the target sequence. In situations where there is al-
ready a gap in the given position, a random posi-
tion is chosen, the previous gap is maintained, and
the new gap is inserted into that position. The algo-
rithm presented better solutions for a set of Ribonu-
clease sequences extracted from the BAliBase dataset,
which are sequences with an identity above 35%. For
other sequences, with smaller identities, such as Cy-
tochrome c, the results are not much better, but still,
the difference between the minimum and maximum
scores is small, which denotes robustness in the solu-
tion.
2.2 Chaotic Step Length Artificial Bee
Colony Algorithms for Protein
Structure Prediction
An important area of bioinformatics, in addition to
pattern recognition and multiple alignment of se-
quences, is the area of prediction of the protein struc-
ture. Originally, nuclear magnetic resonance and X-
ray crystallography techniques were used to deter-
mine the structure of the protein. However, these ap-
proaches require a costly laboratory equipment and
also consume a lot of time.
The work of Saxena et al. (2020) is in the predic-
tion of the protein structure field through a computa-
tional physical model. In general, the physical models
of protein prediction are constructed in two phases:
initially, a model with unknown energy is created and
optimization functions are applied on this model to
minimize protein-free energy.
To build and optimize free energy models, meta-
heuristics are often used, one of these meta-heuristics
is the Artificial Bee Colony. In the work of Saxena
et al. (2020), the hypothesis of applying chaotic func-
tions in a step of the algorithm Artificial Bee Colony
(ABC) is validated for best results. Originally, the
ABC algorithm updates bee velocity using the equa-
tion 1, where Φ is a randomly generated number in the
interval [−1, 1]. Saxena’s proposition is to replace the
random value with a method called Chaotic Length
Separator. The modified function is represented ac-
cording to the equation 2.
υ
i j
= x
i j
+ φ(x
best j
− x
i j
) (1)
υ
i j
= x
i j
+CLS
t
(x
best j
− x
i j
) (2)
To calculate Chaotic Length Separator, ten chaotic
map functions were chosen. The method were now
named Enhance Chaotic Artificial Bee colony and
for each function an indice was assigned. Some of
them are: Chebyshev chaotic (ECABC1), Sinusoidal
chaotic (ECABC9) and Tent chaotic (ECABC10).
After computing the functions, the result is normal-
ized using the equation 3, in the interval [0.2,1
e−10
]
where, t denotes the current iteration and T the maxi-
mum number of iterations.
ICEIS 2025 - 27th International Conference on Enterprise Information Systems
238

Table 1: Results of chaotic PSO optimization.
Identity Sequence Average Maximum Minimum
< 25% SH3 0.5971 0.9461 0.4559
twitchin 0.4721 0.5248 0.4215
20% - 40% SH2 0.6235 0.6807 0.5723
Cytochrome c 0.5346 0.5731 0.4822
> 35% Ribonuclease 0.8256 0.8761 0.7778
immunophilin c 0.5146 0.5739 0.4611
N
m
(t) = N
max
m
−
N
max
m
− N
min
m
T
∗t (3)
The results pointed out by the study indicate bet-
ter efficacy in the conversion of the ABC algorithm
with the use of chaotic maps. In addition, this ap-
proach, with ten distinct functions, allowed exploring
and highlighting the differences and behavior of the
algorithm with its use of each of them.
3 THE PROPOSED METHOD
The present work deals with the refinement of mul-
tiple alignment of sequences through a hybridization
of techniques, aiming to obtain more biologically rel-
evant results. To achieve this objective, several ap-
proaches have been implemented and the main one,
which is at the heart of the proposed method, is the
ant colony optimization algorithm. It is through this
method that other approaches, such as the realign-
ment of sequence fragments and the application of the
chaotic method, are applied.
This section is subdivided into 5 parts: Method
Overview, Ant Colony Optimization, Multiple Se-
quence Alignment, Subsequence Realignment, and fi-
nally, Chaotic Jump. In the overview, a pseudocode of
the method is presented to facilitate understanding. In
the Ant Colony Optimization section, the ant colony
optimization algorithm, its modeling for the sequence
alignment problem and the parameters used will be
detailed.
3.1 Method Overview
The proposed method is to apply Ant colony opti-
mization to refine the multiple alignment of sequences
constructed by the KAlign and Omega Clustal tools.
To achieve this goal, this method captures the initial
alignment produced and performs refinement using
ACO optimization, recomputate the pair-by-par align-
ment, and realigns with the Center Star technique.
When the alignment resulting from refinement is
better than the previous one, this alignment is stored
as a possible final solution, otherwise a counter is in-
cremented. If the counter obtains a predetermined
threshold, the algorithm enters a realignment phase.
At this stage, a position of the sequences is chosen us-
ing chaotic draw, where the chaotic jump is applied at
an initially random value. Only the first random val-
ues drawn are used, because in the next occurrences,
the value is backfed in the chaotic formula itself to
recalibrate the initial position of the realignment.
The realignment step selects the starting position
and size that will be recalibrated and performs a mul-
tiple alignment of only that piece of the sequences and
then inserts them back into the original positions. Af-
ter this realignment, a new iteration will be performed
with the sequences modified to re-measure the quality
of the alignment performed. This cycle repeats until
the limit of iterations is reached. The Algorithm 1 il-
lustrate these steps that were described in the previous
paragraphs.
Algorithm 1. Overview of the proposed method.
for ant = 1, 2, . . . , N do
for iteration = 1, 2, . . . , N do
Generate initial multiple alignment
(ClustalW and KAlign)
Computing pair-by-par alignments
Run Center Star for Multiple Alignment of
Sequences
Calculate the score of multiple sequence
alignment
if Multiple alignment better than the previ-
ous one then
Store better alignment
else
Compute execution without improving
if Reached limit of executions without
improving then
Realign fragments of all sequences
end if
end if
end for
end for
Multiple Sequence Alignment Using Ant Colony Optimization with Chaotic Jump
239

3.2 Ant Colony Optimization
The approach of the proposed method, specifically
with regard to ant colony optimization is based on the
work of Amorim (2017). In his work, multiple align-
ment of sequences is improved by mutations in pre-
aligned sequences using a simplified grid to displace
ants and update the pheromone track by this grid. As
ants select the paths by grid, they also deposit the
pheromones on the chosen track, so that the trail that
generates the best biological result will be preferred
and trails with lower results will be discarded.
During the trail offset, when the ant takes a path
in the grid of the top line or infer line, a gap is in-
serted into the unchosen line, if the ant follows the
middle line, no gap is inserted and the two sequences
are not modified. Because of mutations, the result-
ing sequences are previously resized to a size of (2 *
max) positions, where max corresponds to the largest
size sequence and empty spaces are filled with gaps.
This procedure is done only to normalize the size of
the sequences and allow the realignments to fit in the
sequences in their initial positions.
At the beginning of the process, ants use the
ClustalW and KAlign tools to produce an initial ba-
sic alignment. These tools were chosen because their
response time is in the millisecond to seconds and
the quality of the alignment produced is reasonable.
After the construction step of the initial alignment,
the pair-by-par alignment of each sequence is per-
formed, using the meta-heuristic Ant Colony Opti-
mization (ACO) to converge on biologically more rel-
evant alignments.
The equation 4 denotes the probability of transi-
tioning to the grids of the pair-by-par alignment. The
dimension i = (0, 1, 2) corresponds to the path that the
ant will choose, 0 for the top row, 1 for the middle
line, and 2 for the bottom row. Dimension j = 1..N
th
corresponds to the residue or amino acid of the se-
quence, and the dimension k = 1..T corresponds to
the number of the initial sequence of the pair-by-pair
alignment, where T is the total number of sequences.
The value of eta is set at 1, since because the simpli-
fied grid has only 3 options, this value does not inter-
fere with convergence. The value of al pha and beta
are method parameters and their values will be set by
the user. All parameter values and their choice criteria
will be presented in section 4.
P
z
i jk
=
τ
i jk
(t)
α
η
β
i jk
∑
τ
i jk
(t)
α
η
β
i jk
(4)
After each pair is aligned, a score is computed
for the alignment. This score is used to update the
intensity of the pheromone track, which serves as
a guide for ants to decide between one path or an-
other. The equation representing the evaporation of
path pheromones can be observed in 5. In this equa-
tion, p is a parameter in the [0, 1] range and represents
the evaporation rate. The value of ∆τ
i jk
is defined by
the equation 6, in which Q and MaxScore are param-
eters for the algorithm, and Score
z
is the calculated
score for the newly aligned sequence pair and is de-
fined by the 7 equation.
τ
i jk
(t + 1) = (1 − p) ∗ τ
i jk
(t) + ∆τ
i jk
(5)
∆(τ
i jk
) =
Q ·
Score
z
+ MaxScore
2
, if the ant z
transit through the grid (i, j, k)
0, if the ant z
do not transit through the grid (i, j, k)
(6)
Score(a, b) =
len
∑
i=1
α(A
′
[i], B
′
[i]) (7)
This pair-by-par alignment process is repeated for
all sequences, and upon completion, a 2-dimensional
array is generated. This array has 2 dimensions to al-
low storing all alignments against all sequences. In
addition, another resulting vector stores the cumula-
tive score of all alignments for each sequence. Both
artifacts are used in the next step, which is multiple
alignment of sequences.
3.3 Multiple Sequence Alignment
Among several multiple alignment methods, this
work uses the Center Star Zou et al. (2015) method.
This method consists of selecting the sequence
that scored the highest, starting multiple alignment
through it, and progressively inserting the other se-
quences, taking into account the pair-by-par align-
ment matrix.
In Figure 1, we observe the process of construct-
ing multiple alignment by this technique, this process
is divided into three steps, with a left enumeration in-
dicating each step. In step 1, the sequences are to be
aligned. In step 2, a representation of the par-to-par
alignment score can be observed, the objective is to
find the sequence that obtains the highest score in this
matrix, for this each sequence is positioned in the ma-
trix with a score that corresponds to the result of the
pair-by-par alignment performed for each of the other
sequences present. And finally, in step 3, the s1 se-
quence was selected as center star and the alignment
ICEIS 2025 - 27th International Conference on Enterprise Information Systems
240
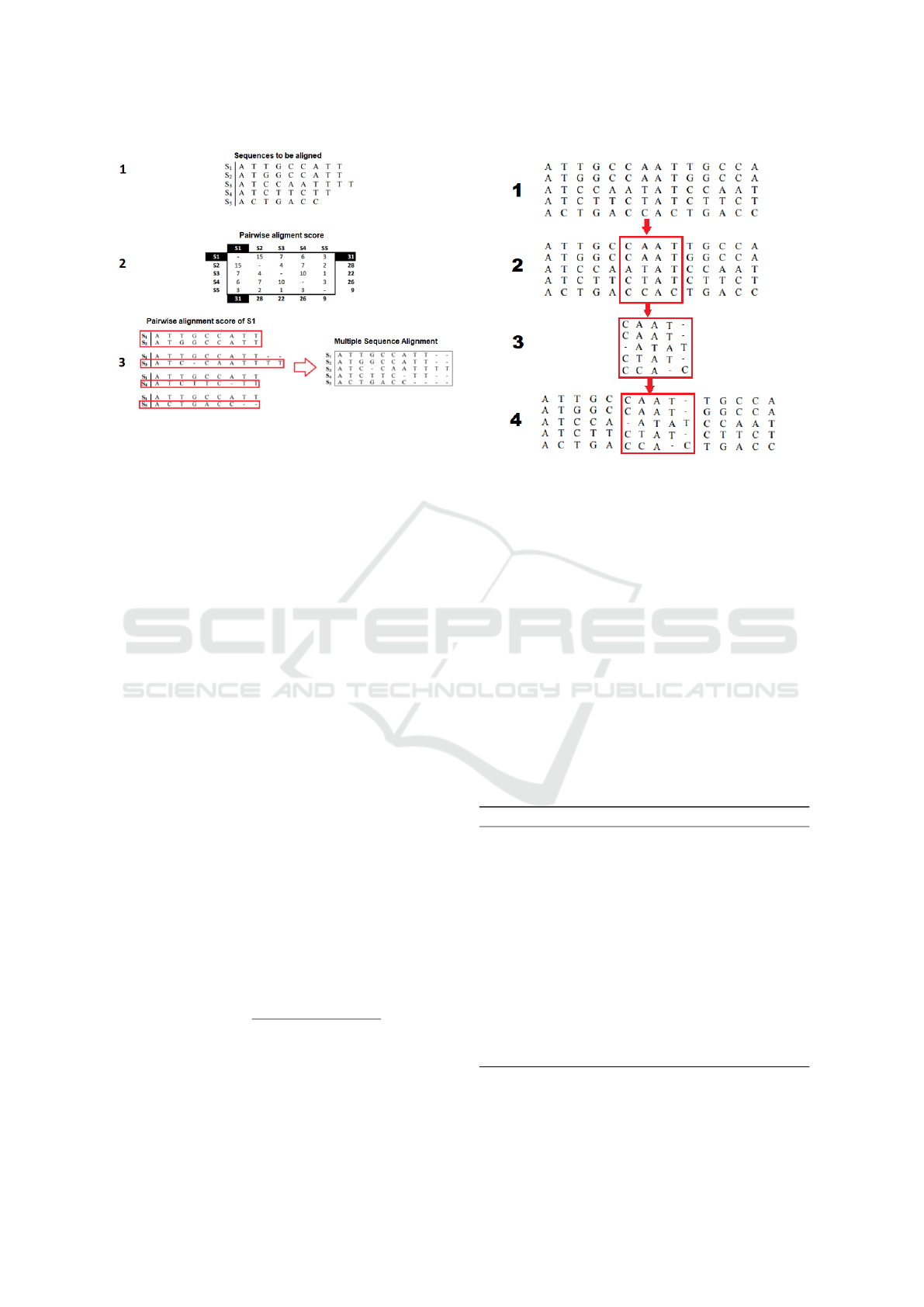
Figure 1: Multiple sequence alignment by Center Star algo-
rithm - Source: Adapted of Amorim (2017).
is constructed from the pair-by-par alignments with
that sequence.
To calculate the score of multiple sequence align-
ment, this work used the weighted sum-of-pairs
(WSP) Rubio-Largo et al. (2015) function. This is
the objective function that is maximized in the heuris-
tics developed in the present work. In the equation 8,
which defines the WSP function, AL is the size of the
aligned sequences and k is the total sequences. There-
fore, l corresponds to each residual or position of the
sequence and s
′
i
corresponds to the sequence resulting
from the multiple alignment. The SP(l) function is
defined by the equation 9.
WSP(S
′
) =
AL
∑
l=1
SP(l) −
k
∑
i=1
AGP(s
′
i
) (8)
SP(l) =
k−1
∑
i=1
k
∑
j=1
W
i j
× δ(s
′
i,l
, s
′
j,l
) (9)
In 9, delta corresponds to some replacement ar-
ray (BLOSUM, PAM). The function of this matrix
in the equation is to weigh the weight of replacing
one amino acid with another. In the present work,
we used the BLOSUM62 Eddy (2004) matrix. The
W
i, j
(a, b) function is defined by the equation refdis-
tance, in which LD(s
i
, s
j
) corresponds to the distance
of Levenshtein and s
i
and s
j
correspond to the un-
aligned sequences.
W
i, j
(a, b) = 1 −
LD(s
i
, s
j
)
max(len(s
i
), len(s
j
))
(10)
Finally, in the equation 11, you have the penalty
function for gaps, where g
0
is the weight for each
opening of a new gap and g
e
is the weight applied to
the total of gaps consecutive after each new opening.
Figure 2: Example of the realignment phase.
AGP(s
′
i
) = (g
0
× N
gaps
) + (g
e
× N
spaces
) (11)
3.4 Subsequence Realignment
When the algorithm performs a certain number of iter-
ations and the quality of multiple sequence alignment
does not improve, a realignment step is performed on
subsequences of all sequences involved in multiple
alignment. The value of the number of iterations is
the limiting factor for selecting the position in the se-
quence, in which the subsequences will be extracted,
in which the logistic map function is applied. The re-
sult of this function is translated to a position in the se-
quence and alignment is performed using the KAlign
tool. This realignment phase can be observed through
the algorithm 2:
Algorithm 2. Realignment algorithm overview.
Calculate realignment limits
Extract fragments the same size from all sequences
Generate new file with only fragments of the origi-
nal sequences
Run KAlign
Get sequences aligned by KAlign
Inject aligned sequences into the originally ex-
tracted ranges
Multiple Sequence Alignment Using Ant Colony Optimization with Chaotic Jump
241

3.5 Chaotic Jump
Chaotic Jump is used in the method proposed in the
sequence position selection step to perform realign-
ment. First, the logistic map function was defined ac-
cording to the equation 12. For the present study, the
value of α = 4 was used, because with this value the
result of the function, for any value of x, will always
occur in the range [0, 1]. Because this function ex-
pects a value to calculate the next, the initial value is
randomly generated in the same range.
δ
p
= x
n+1
= αx
n
(1 − x
n
) (12)
In the 3 algorithm, how the value of delta
p
is
transformed into a sequence position and how the
block size that will be realigned is determined. In
the first two lines, the minimum and maximum lim-
its of the block size that will be realigned are calcu-
lated respectively. In the third line, the fact size of the
block is calculated randomly in the range of the previ-
ously defined limits. Therefore, the limit size is set for
the draw of the position, to prevent the drawn num-
ber from exceeding the length of the sequences. After
that, the start and end positions are calculated, and the
extraction process takes place as demonstrated in the
previous section. The values of P
textmin
and P
textmax
are algorithm parameters and initialized by default at
5 and 25 of the total sequence size, respectively.
Algorithm 3. Algorithm used to determine realignment po-
sitions and sizes.
Block
min
= P
min
∗ T
seq
Block
max
= P
max
∗ T
seq
Block
size
= Rand(Block
min
, Block
max
)
T
limit
= T
seq
− Block
size
P
initial
= δ
p
∗ T
limit
P
f inal
= P
initial
+ Block
size
4 RESULTS AND DISCUSSION
All tests of this work were run under a computer
with Windows Operating System(R) 10 Enterprise, 16
GB RAM, an Intel(R) Core(TM) i7-8665U CPU @
1.90GHz 2.11GHz with 4 cores and 8 threads and a
240GB SSD.
4.1 Parameters Used in the Present
Work
The parameters used for ACO, WSP and Realignment
methods are shown in table 2.
In order to compare the efficiency of the proposed
method, the same set of sequences was, in paral-
lel, aligned using the MSA-GA, KAlign and Clustal
Omega tool. The parameters used by the MSA-GA
tool are in Table 3:
All tests performed were performed on the BAl-
iBase 3 benchmark. 10 random regions of all BAl-
iBase families were selected, and the tests were re-
peated at least 3 times in each tool to obtain the mean
and standard deviation of the metrics. To measure the
quality of alignment, the QScore tool was used, which
is a customized version of the BAliScore tool to ac-
cept the .fasta file format.
The comparison of the alignments made by the
tools are shown in tables 4, 5, 6, 7 and 8. In the ta-
bles’ columns are presented: the region of BaliBase
(column Region), the method used (column Method),
the average score of Q (column Avg. Q), the maxi-
mum score of TC (column Max TC) and the standard
deviation of Q score (column SD Q).
5 CONCLUSIONS
In this work, a method for multiple alignment of
sequences using Ant Colony Optimization was pro-
posed and implemented in conjunction with a chaotic
state approach. To validate the results, the propo-
nent made comparations of the new method with
three other methods also implemented in tools, called
MSA-GA, KAlign and Clustal Omega.
Through the analysis of the results and the
comparative table we can observe that the proposed
method showed better results, regarding the quality of
the alignment produced, in more than fifty percent of
the executions, for all regions used in the benchmark.
In tables 4, 5, 6, 7 and 8, it can be observed that
the value of the ACO method is at least as good as
the best alignment between the Omega clustal and
KAlgin tools. This behavior is expected, since to
perform the realignments, these tools are used to gen-
erate an initial alignment. And because the proposed
method is a refinement of multiple alignment, the
quality will be at least as good as the alignment pro-
duced by the initial alignment. When observing the
values of the standard deviations, it is also perceived
that the method offers a robust solution, because in
more than half of the cases, the result value always
ICEIS 2025 - 27th International Conference on Enterprise Information Systems
242
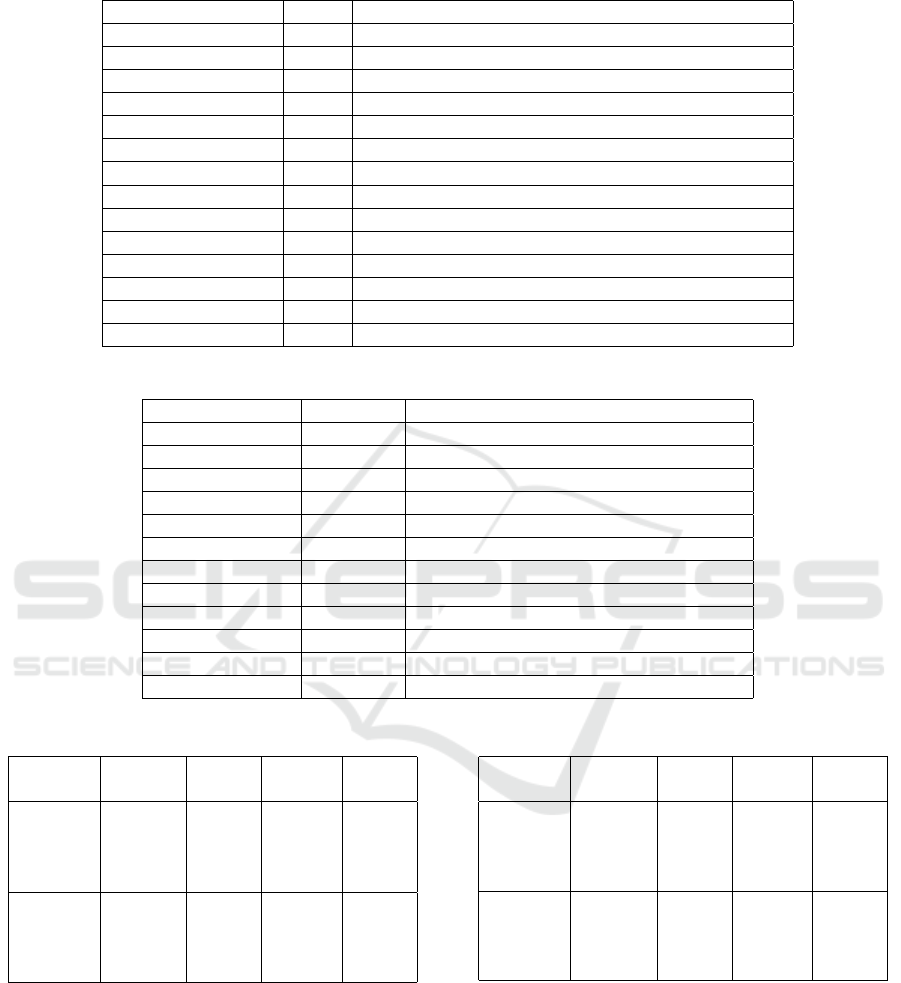
Table 2: Parameters for the ACO, WSP and Realignment methods.
Name Value Description
NumberOfAnts 20 Amount of ants used
NotImprovedLimit 10 Minimum number of iterations
NumberOfIterations 300 Maximum number of iterations per ant
α 1.0 Value of alpha to ACO
β 5.0 Value of beta to ACO
Q
0
0.3 Value of Q0 to ACO
RO 0.2 Value of RO to ACO
Q 0.001 Value of Q to ACO
τ
0
10 Initial pheromone value to ACO
MaxScore 200.0 Value of MaxScore to ACO
G
e
0.85 Value of Gap Extension penalty for the WSP function
G
0
6 Value of Gap Open penalty for the WSP function
MinFragmentPerc 0.05 Minimum percent of the sequence block size
MaxFragmentPerc 0.25 Maximum percent of the sequence block size
Table 3: Parameters used in the MSA-GA tool.
Name Value Description
PopulationSize 1000 Population size
Generations 10000 Number of Generations
BlockMutation 0.01 Mutation rate
GapExtMutation 0.05 Gap Extension Mutation
GapRedMutation 0.05 Gap Reduction Mutation
HorCrossover 0.8 Horizontal Cross-over rate
VerCrossover 0.8 Vertical Cross-over rate
HorVerRation 0.5 Horizontal/Vertical Ratio
TournamentSize 2 Tournament Size
OffSet 0 Offset value
MaxSeqSize 1.2 Maximum size of the sequence alignment
Matrix Blosum62 Substitution matrix
Table 4: Results for the regions BB11001 and BB11010.
Region Method Avg.
Q
Max
TC
SD Q
BB11001 ACO 0.969 0.943 0.05
BB11001 ClustalO 0.907 0.83 0
BB11001 KAlign 0.874 0.727 0
BB11001 MSAGA 0.728 0.5 0.02
BB11010 ACO 0.299 0.175 0.02
BB11010 ClustalO 0.287 0.14 0
BB11010 KAlign 0.283 0.148 0
BB11010 MSAGA 0.141 0.00388 0.13
generates the same value and therefore the devi-
ation is zero.
As a future works, we intend to investigate the
improvement to this method in order to reduce the
execution time. Since the Ant Colony Optimization
allows to share minimum state between ants, a par-
allel approach would be viable and recommended to
achieve this goal.
Table 5: Results for the regions BB12012 and BB12022.
Region Method Avg.
Q
Max
TC
SD Q
BB12012 ACO 0.564 0.42 0.05
BB12012 ClustalO 0.558 0.41 0
BB12012 KAlign 0.538 0.387 0
BB12012 MSAGA 0.138 0.0173 0.09
BB12022 ACO 0.783 0.558 0
BB12022 ClustalO 0.772 0.532 0
BB12022 KAlign 0.719 0.468 0
BB12022 MSAGA 0.628 0.374 0.03
ACKNOWLEDGEMENTS
The authors would like to thank S
˜
ao Paulo Research
Foundation (FAPESP) for the financial support, un-
der grants, 20/08615-8, 23/13399-0, 23/13576-0,
23/13610-3, and Coordenac
˜
ao de Aperfeic¸oamento
de Pessoal de N
´
ıvel Superior - Brasil (CAPES) for
the partial financial support.
Multiple Sequence Alignment Using Ant Colony Optimization with Chaotic Jump
243
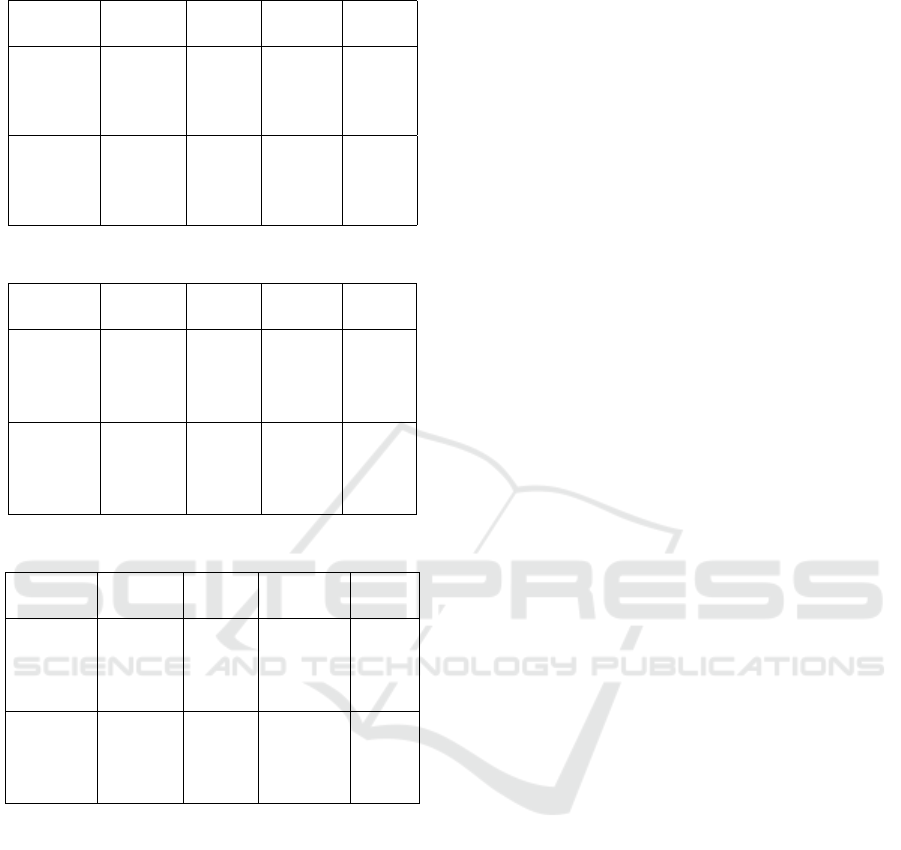
Table 6: Results for the regions BB20010 and BB20030.
Region Method Avg.
Q
Max
TC
SD Q
BB20010 ACO 0.852 0.535 0
BB20010 ClustalO 0.852 0.535 0
BB20010 KAlign 0.826 0.44 0
BB20010 MSAGA 0.298 0.00152 0.08
BB20030 ACO 0.895 0.0955 0
BB20030 ClustalO 0.883 0.0892 0
BB20030 KAlign 0.781 0.052 0
BB20030 MSAGA 0.441 0.024 0.11
Table 7: Results for the regions BB30016 and BB30024.
Region Method Avg.
Q
Max
TC
SD Q
BB30016 ACO 0.414 0.0641 0
BB30016 ClustalO 0.414 0.0641 0
BB30016 KAlign 0.311 0.0148 0
BB30016 MSAGA 0.0907 0.0148 0
BB30024 ACO 0.726 0.448 0
BB30024 ClustalO 0.726 0.448 0
BB30024 KAlign 0.706 0.392 0
BB30024 MSAGA 0.0171 0 0.12
Table 8: Results for the regions BB40011 and BB50014.
Region Method Avg.
Q
Max TC SD Q
BB40011 ACO 0.363 0 0
BB40011 ClustalO 0.358 0.000569 0
BB40011 KAlign 0.363 0 0
BB40011 MSAGA 0.00291 0 0.09
BB50014 ACO 0.811 0.398 0.02
BB50014 ClustalO 0.803 0.395 0
BB50014 KAlign 0.756 0.27 0
BB50014 MSAGA 0.231 0.0184 0.01
REFERENCES
Amorim, A. R. (2017). Alinhamento m
´
ultiplo de
sequ
ˆ
encias utilizando algoritmo gen
´
etico multifunc¸
˜
ao
e col
ˆ
onia de formigas. Master’s thesis, Universidade
Estadual Paulista (UNESP).
Eddy, S. R. (2004). Where did the blosum62 alignment
score matrix come from? Nature biotechnology,
22(8):1035–1036.
Gomes, V. Z., Andrade, M. C., Amorim, A. R., and Zafalon,
G. F. D. (2022). A hybrid genetic algorithm using pro-
gressive alignment and consistency based approach
for multiple sequence alignments. In Proceedings of
the 24th International Conference on Enterprise In-
formation Systems - Volume 2: ICEIS, pages 167–174.
INSTICC, SciTePress.
Lassmann, T. (2019). Kalign 3: multiple sequence align-
ment of large datasets. Bioinformatics, 36(6):1928–
1929.
Lee, Z.-J., Su, S.-F., Chuang, C.-C., and Liu, K.-H. (2008).
Genetic algorithm with ant colony optimization (ga-
aco) for multiple sequence alignment. Applied Soft
Computing, 8(1):55–78.
Lei, X.-j., Sun, J.-j., and Ma, Q.-z. (2009). Multiple se-
quence alignment based on chaotic pso. In Interna-
tional Symposium on Intelligence Computation and
Applications, pages 351–360. Springer.
Prosdocimi, F., Coutinho, G., Ninnecw, E., Silva, A. F.,
dos Reis, A. N., Martins, A. C., dos Santos, A. C. F.,
J
´
unior, A. N., and Camargo Filho, F. (2002). Bioin-
form
´
atica: manual do usu
´
ario. Biotecnologia Ci
ˆ
encia
& Desenvolvimento, 29:12–25.
Rubio-Largo,
´
A., Vega-Rodr
´
ıguez, M. A., and Gonz
´
alez-
´
Alvarez, D. L. (2015). A hybrid multiobjective
memetic metaheuristic for multiple sequence align-
ment. IEEE Transactions on Evolutionary Computa-
tion, 20(4):499–514.
Rubio-Largo,
´
A., Vega-Rodr
´
ıguez, M. A., and Gonz
´
alez-
´
Alvarez, D. L. (2016). Hybrid multiobjective artificial
bee colony for multiple sequence alignment. Applied
Soft Computing, 41:157–168.
Saxena, A., Shekhawat, S., Sharma, A., Sharma, H., and
Kumar, R. (2020). Chaotic step length artificial bee
colony algorithms for protein structure prediction.
Journal of Interdisciplinary Mathematics, 23(2):617–
629.
Sievers, F. and Higgins, D. G. (2014). Clustal omega, ac-
curate alignment of very large numbers of sequences.
In Multiple sequence alignment methods, pages 105–
116. Springer.
Zafalon, G. F. D. (2009). Algoritmos de alinhamento
m
´
ultiplo e t
´
ecnicas de otimizac¸ao para esses algorit-
mos utilizando Ant Colony. PhD thesis, Universidade
Estadual Paulista (UNESP), R. Crist
´
ov
˜
ao Colombo,
2265 - S
˜
ao Jos
´
e do Rio Preto - SP, Brasil.
Zafalon, G. F. D. Z., Gomes, V. Z., Amorim, A., and
Val
ˆ
encio, C. R. (2021). A hybrid approach using pro-
gressive and genetic algorithms for improvements in
multiple sequence alignments. In Proceedings of the
23rd International Conference on Enterprise Informa-
tion Systems - Volume 2: ICEIS, pages 384–391. IN-
STICC, SciTePress.
Zou, Q., Hu, Q., Guo, M., and Wang, G. (2015). Halign:
Fast multiple similar dna/rna sequence alignment
based on the centre star strategy. Bioinformatics,
31(15):2475–2481.
ICEIS 2025 - 27th International Conference on Enterprise Information Systems
244
