
Progress in CYP Enzymes Mechanisms of Induction and Its
Applications
Zhanxu Meng
Tai 'an No. 1 Middle School, Tai 'an, Shandong Province, China
Keywords: Cytochrome P450 (CYP) Enzymes, Induction Mechanisms, Drug Metabolism, In vitro, In vivo.
Abstract: Cytochrome P450 (CYP) enzymes are pivotal in the metabolic processing of drugs, and their modulation via
induction pathways plays an important role in the therapeutic efficacy, safety, and drug interaction profile.
This comprehensive review delineates three focal points concerning CYP enzyme induction. The first domain
scrutinizes the accuracy of in vitro methodologies—predominantly cryopreserved primary human hepatocytes
(PHH) and Human HepaRG cells—in forecasting the in vivo dynamics of CYP enzymes. The second segment
elucidates novel induction paradigms, highlighting the enduring induction of CYP1A enzymes via 3-
methylcholanthrene (MC) in murine models and the atypical induction pathways that involve YAP/TEAD
perturbation and consequent hepatocyte dedifferentiation. The terminal section evaluates the application of
these findings in clinical settings, discussing the kinetic profiling of CYP3A modulation and the transposition
of in vitro CYP repression to actual drug-drug interaction scenarios. The synthesis of these facets contributes
to an enriched understanding of CYP induction mechanisms and their ramifications for drug discovery and
tailored therapeutic approaches. Furthermore, the appraisal accentuates the reliability and pertinence of
cryopreserved PHH and HepaRG cells as in vitro proxies for human CYP enzyme induction studies,
potentially informing regulatory risk evaluation and elucidating drug metabolism and nuclear receptor-
mediated regulatory anomalies in biochemical pathways.
1 INTRODUCTION
Cytochrome P450 (CYP) enzymes play a critical role
in drug metabolism, facilitating the biotransformation
of a wide range of endogenous and exogenous
compounds within the body (Pelkonen et al. 2008).
The regulation of CYP expression is essential for
maintaining physiological balance and ensuring
efficient drug metabolism (Li et al., 2019). Induction
represents a primary mechanism through which CYP
expression levels are modulated, whereby various
xenobiotics and endogenous signalling molecules can
upregulate these enzymes (Li et al., 2019). When the
drug is cleared from the body more quickly, it leads
to lower levels of the drug in the body, which can
reduce its effectiveness. An example of this is when
rifampin is taken alongside sulfonylureas, resulting in
decreased levels of sulfonylureas in the blood and a
diminished ability to lower blood glucose. This
decrease in effectiveness may be caused by the
induction of CYP2C9 enzyme by rifampin. Another
factor contributing to the decreased drug
concentrations and effectiveness could be the
induction of P-glycoprotein by rifampin.
CYPenzymes play a critical role in drug
metabolism, with induction and inhibition as key
modulatory mechanisms. Induction of these enzymes
is characterized by a time-dependent augmentation in
enzyme levels, necessitating a period of adaptation to
stabilize at a new homeostasis (Lin, 2006).
Contrasting the immediate effect of CYP inhibition,
induction involves intricate genetic regulation
leading to an increase in enzyme synthesis.
Activation of CYP enzymes within the families 1 to 3
is a complex process governed by three primary
pathways, responding to exogenous compounds
(Pelkonen et al. 2008). Under basal conditions, these
receptors are sequestered in the cytoplasm. It is bound
to heat shock protein 90 (Hsp90). Ligand binding
instigates a conformational alteration, prompting
dissociation from Hsp90, receptor activation, and
nuclear translocation, thereby kick-starting gene
transcription. Beyond these classical receptor-
mediated mechanisms, CYP enzyme induction also
encompasses pathways such as the direct and indirect
76
Meng, Z.
Progress in CYP Enzymes Mechanisms of Induction and Its Applications.
DOI: 10.5220/0013846300004914
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 2nd International Conference on Renewable Energy and Ecosystem (ICREE 2024), pages 76-86
ISBN: 978-989-758-776-4
Proceedings Copyright © 2025 by SCITEPRESS – Science and Technology Publications, Lda.

glucocorticoid receptor-mediated induction,
highlighting the nuanced regulatory landscape of
CYP enzyme activity (Schuetz et al., 1996 & Pascussi
et al., 2001). Moreover, the modulation of certain
CYP enzymes transcends transcriptional control,
involving post-transcriptional modifications that
stabilize the mRNA and protein, further
complexifying the regulation of CYP enzyme levels
(Chen et al., 2017)
The process of CYP induction holds significant
implications in pharmacology and toxicology,
impacting drug efficacy, safety, and potential
interactions. Understanding the molecular
mechanisms underlying CYP induction is vital for
optimizing drug therapy, predicting drug-drug
interactions (DDIs), and comprehending the effects of
environmental toxins on human health.
This review provides a detailed examination of
CYP induction, highlighting its implications for drug
metabolism and toxicity testing. Anchored by three
primary perspectives, the analysis begins with an
exploration of in vitro methodologies and their role in
predicting in vivo CYP induction, a topic extensively
addressed by Bernasconi et al. in 2019 (Bernasconi et
al., 2019). The translation of in vitro findings to in
vivo contexts is critical, particularly in understanding
how induced CYP activity may affect metabolic
processes. Recent advances have refined these in
vitro techniques, capitalizing on human-based
systems to simulate and study enzyme interactions.
CYP P450 enzymes, key players in xenobiotic
metabolism, are underscored for their expansive
presence and metabolic diversity. They are
instrumental in detoxifying xenobiotics by
metabolizing harmful substances into benign
products or, conversely, converting non-toxic
compounds into harmful metabolites. Beyond
detoxification, these enzymes are essential for
synthesizing various endogenous compounds.
Therefore, the xenobiotic-induced alteration of CYP
enzyme activity has far-reaching effects on metabolic
stability and can precipitate adverse biological
consequences. This review underscores two pivotal
studies utilizing in vitro approaches to forecast drug
interactions and refine clinical outcomes. The first
study by Bernasconi et al. investigates the effects of
tipranavir/ritonavir on enzymatic and transporter
functions (Bernasconi et al., 2019). The second, by
Dumond et al., presents cocktail phenotyping, a novel
strategy for evaluating drug interaction potentials.
Both studies furnish essential insights into the
complex interplay of drug interactions, contributing
to the enhancement of therapeutic protocols (Dumond
et al., 2010). Consequently, the review will critically
analyze and synthesize the findings, emphasizing
their significant contributions to the field of in vitro
prediction methods.
Furthermore, the review delves deeper into novel
mechanisms of CYP enzyme induction, specifically
highlighting the extended induction of CYP1A
enzymes by MC in murine models, a phenomenon
elucidated by Jiang et al. in 2009 (Jiang et al., 2009).
The research has shed light on the persistent
transcriptional activation of promoters associated
with these enzymes. Considering the potent
carcinogenicity of MC, a polycyclic aromatic
hydrocarbon (PAH) prevalent in various
environmental matrices, it's crucial to understand
how its metabolism by CYP enzymes results in
intermediates capable of DNA binding and potential
carcinogenesis.
The induction of CYP1A1 & A2 enzymes by MC
and the eventual decline post-exposure cessation
present an enigmatic aspect of CYP regulation. This
review aims to dissect the mechanisms behind the
lasting transcriptional activation of these CYP
enzymes, which have significant implications for
environmental health. Moreover, the review
addresses a comparative analysis of YAP/TEAD
inhibitors within bidimensional and tridimensionality
primary human hepatocyte cultures conducted by
Oliva-Vilarnau et al. in 2023 (Oliva-Vilarnau et al.,
2023). These inhibitors, initially developed as cancer
therapeutics, have been observed to stimulate CYP
enzymes in 2D hepatocyte cultures. Intriguingly, such
induction is absent in 3D spheroid cultures,
underscoring the importance of considering
alternative induction pathways and adopting
organotypic culture systems in drug development to
predict CYP enzyme modulation.
Clinical trials form the apex of this review,
particularly the meticulous quantification of CYP3A
modulation dynamics through continuous midazolam
(MDZ) infusion and the correlation of in vitro P450
downregulation with in vivo DDIs, especially
regarding 13-cis-Retinoic Acid (13cisRA) (Li et al.,
2019 & Stevison et al., 2019). The time-dependent
modulation of CYP enzymes is critical in the design
of DDI studies, which in turn influences the safe and
efficacious application of drugs metabolized by
CYP3A.
This compendium of studies coalesces into a
comprehensive understanding of CYP enzyme
induction, from experimental exploration to clinical
relevance, imparting vital insights that have the
potential to refine drug development and pave the
way for tailored therapeutic approaches in
personalized medicine.
Progress in CYP Enzymes Mechanisms of Induction and Its Applications
77

2 USING IN VITRO
METHODS FOR
PREDICTING IN VIVO
BEHAVIOUR
In vitro methodologies represent a cornerstone in the
preclinical assessment of CYP enzyme expression
upon exposure to pharmaceuticals. These
methodologies utilize various hepatocyte-based
models as proxies to the in vivo environment, offering
a simplified yet controlled setting to circumvent the
complexities and ethical considerations of animal
testing.
According to the Food and Drug Administration
(FDA) in U.S., a spectrum of in vitro hepatic models
has been established. This suite includes fresh and
cryopreserved primary hepatocytes, hepatocytes with
stable or transient transfections, hepatic cell lines, and
assays utilizing reporter genes. These platforms
enable pharmaceutical entities to gauge the induction
potential of new compounds, aligning their evaluation
processes with the regulatory framework set forth by
the FDA. A compound is flagged for further clinical
drug-drug interaction studies and in vivo scrutiny if it
elicits a CYP enzyme induction surpassing 40%
relative to a positive control, as per FDA guidelines.
Molecules demonstrating significant induction
propensities might be withdrawn from the
development pipeline to pre-empt adverse drug
interactions (Ghosh et al., 2023).
An initial high-throughput screen can detect
enhanced activation of nuclear receptors leading to
upregulated CYP enzyme synthesis. One approach
involves coalescing hepatoma cells with a CYP3A4
promoter region and a luciferase reporter or using a
human pregnane X receptor (PXR) coupled with the
CYP3A4-luciferase construct. Given CYP3A4's
susceptibility to PXR-mediated induction, regulatory
protocols advocate for its in vitro examination during
early drug development. A negative outcome for
CYP3A4 can generally rule out the induction potential
for CYP2C, as PXR also governs this enzyme. Hepatic
and immortalized cell lines that maintain hepatocyte
characteristics and deliver reproducible findings are
integral to the pharmaceutical industry. Among these,
the HepaRG and Fa2N-4 cell lines are frequently used,
with mRNA measurement serving as an accepted
endpoint for induction in immortalized cells.
Hepatoma lines, like HepG2, HepaRG, and BC2, see
extensive application within industry settings (Ghosh
et al., 2023 & Ingelman-Sundberg, 2004).
Primary hepatocytes are endorsed by industry and
regulatory bodies due to their preservation of in vivo-
like CYP metabolism post-isolation. For prolonged
phenotypic stability, 3D cultures are preferred over 2D
monolayers. With primary human hepatocyte cultures,
CYP mRNA, protein levels, and microsomal activity
can be accurately quantified. Furthermore, to address
inter-individual metabolic variance, hepatocyte
cultures sourced from multiple donors are utilized
(Ghosh et al., 2023).
Despite their merits, the application of primary or
cryopreserved hepatocytes is constrained by
challenges such as limited availability, potential loss
of enzymatic function due to cryopreservation, single-
receptor pathway analysis limitations, and variability
across different batches. As a result, immortalized and
hepatic cell lines have surfaced as practical
substitutes. These alternatives furnish manifold
benefits: they facilitate the examination of multiple
receptor-mediated mechanisms, offer an inexhaustible
supply via cell propagation, and ensure uniform
inducer responses. Current regulatory guidance also
transitions the focus from enzyme activity to mRNA
expression as the definitive endpoint for in vitro
assays. This shift underscores mRNA levels as a more
dependable metric for gauging CYP induction (Ghosh
et al., 2023).
2.1 Vitro Methods: Human
Cytochrome P450 Enzyme
Induction
The 2019 research by Bernasconi et al. pursued the
validation of two in vitro methodologies designed to
assess chemical compounds' propensity to activate
CYP enzymes—particularly CYP1A2, CYP2B6, and
CYP3A4 (Bernasconi et al., 2019). These
investigations employed two cellular models:
cryopreserved PHH and HepaRG human cells.
The choice of cryopreserved PHH was predicated
on their diverse array of native drug-processing
enzymes and necessary cofactors, establishing them
as a versatile tool for exploring toxicokinetic and
toxicodynamic phenomena. PHH stands as a robust in
vitro proxy for the human liver's metabolic processes.
In parallel, HepaRG cells are recognized for their
hepatic-like functionality, mirroring the metabolic
processes of actual human hepatocytes. This includes
the synthesis of key liver enzymes, the operation of
nuclear receptors, and the facilitation of xenobiotic
transporters. The criterion for validating these
models' metabolic competence was the induction of
CYP enzymes—a vital marker for evaluating the
cellular expression machinery's integrity and
functionality.
ICREE 2024 - International Conference on Renewable Energy and Ecosystem
78
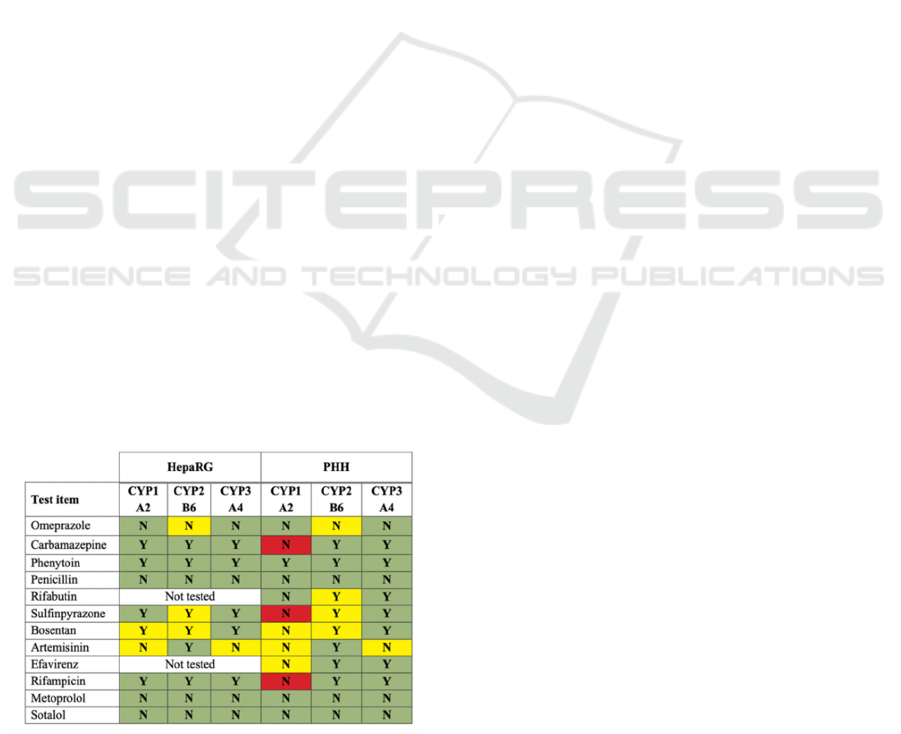
The methodology detailed by the authors
leveraged differentiated HepaRG cells, preserved
through cryopreservation, to monitor the stimulation
of specific CYP enzymes. This process involved the
application of chemical agents to the cells and the
subsequent measurement of CYP1A2, CYP2B6, and
CYP3A enzyme induction via metabolite
quantification using advanced liquid
chromatography/mass spectrometry techniques.
Results indicated that CYP enzyme induction is
not merely a sensitive marker for protein synthesis
but is also a critical parameter for determining hepatic
metabolic capacity. Prior validations have confirmed
the reliability of both PHH and HepaRG cells in
gauging.the functional induction of the specified CYP
enzymes. The robustness of these results is further
corroborated by ring trial data demonstrating
consistent reproducibility across different
laboratories. These findings underscore the methods'
translatability and applicability across research
facilities equipped with cell culture and analytical
chemistry capabilities. Moreover, these in vitro
techniques have been corroborated for their
predictive accuracy regarding in vivo CYP enzyme
induction by various chemicals, evidenced by their
correct identification of reference inducers and the
successful prediction of in vivo human CYP
induction for a majority of the chemicals tested.
Ultimately, the decision to employ PHH or HepaRG
cells hinges on the specific evaluative needs of the
chemical assessment in question.
However, there have been discrepancies in
prediction for certain chemicals in PHH.
Carbamazepine, sulfinpyrazone, and rifampicin in
PHH did not align with in vivo human CYP induction
as expected (Table 2). This could be attributed to the
absence of sufficient human data or variability in the
hepatocyte batches used in the studies.
Figure 1: a comparative analysis of the predictive
capabilities of HepaRG cells and PHHin evaluating the
induction of CYP1A2, CYP2B6, and CYP3A4 (Bernasconi
et al., 2019).
The current validation study supports the
reliability and relevance of cryopreserved PHH and
cryopreserved HepaRG cells as in vitro tools for
assessing human CYP enzyme induction. These in
vitro methodologies are proving valuable in
regulatory risk assessment, offering insights into
metabolic processes, thyroid disruption, and nuclear-
receptor-mediated changes in biochemical pathways.
Utilizing these approaches helps researchers
understand the potential risks of xenobiotic exposure,
guiding the development of more accurate and
effective risk assessment frameworks.
2.2 CYP450 & P-Glycoprotein
Interactions’ Prediction
The Dumond et al. conducted a comprehensive study
to evaluate the potential for DDIs in the combination
therapy of tipranavir/ritonavir (TPV/r), used to treat
HIV-1 infections. Tipranavir, a protease inhibitor
with high efficacy against drug-resistant strains, is
known to induce certain cytochrome P450 enzymes,
particularly CYP3A (Dumond et al., 2010). To offset
this, it is coadministered with ritonavir (RTV), a
CYP3A inhibitor that helps sustain therapeutic levels
of tipranavir. This combination, however,
complicates the accurate prediction of drug
interactions because of its varying effects on
metabolic enzymes and transporters, a challenge that
this study aimed to address.
The researchers utilized a modified cocktail
phenotyping approach to evaluate potential drug
interactions resulting from the coadministration of
TPV and RTV. The study employed the caffeine,
warfarin, omeprazole, dextromethorphan,
intravenous MDZ, and vitamin K, to gauge the
activity of key hepatic and intestinal proteins and
explore the impact of TPV/RTV on them.
In vitro analyses had indicated that the TPV/RTV
combination inhibits several CYP enzymes, such as
CYP3A4, CYP1A2, CYP2C19, and CYP2D6. Yet, in
clinical settings with HIV-1-infected patients on
TPV/RTV and other CYP3A4 substrates, a decrease
in exposure to coadministered protease inhibitors was
observed, contrary to expectations. The study aimed
to resolve this inconsistency by investigating how
TPV/RTV influences various CYP enzymes and by
exploring genotype-phenotype correlations and the
genetic factors that contribute to drug interactions.
The recent investigation has furnished a
multifaceted analysis of the drug-drug interaction
potential attributed to tipranavir/ritonavir
(TPV/RTV) via a comprehensive utilization of probe
substrates and extensive genotyping. This
Progress in CYP Enzymes Mechanisms of Induction and Its Applications
79

encompasses the genotypic characterization of
critical CYP isoforms and P-glycoprotein (P-gp),
endeavoring to articulate the interaction landscape at
various pharmacokinetic junctures—baseline,
subsequent to acute exposure, and upon achievement
of a steady state. Study participants were
administered a cocktail of probe substrates and
digoxin, both via oral and intravenous routes, across
three distinct stages to delineate the
pharmacodynamic profile: baseline, after the triad of
initial TPV/r dosages, and at the steady-state
concentration.
Empirical results divulged that an inaugural dose
of TPV/r exerts negligible modulation on CYP1A2
and CYP2C9 activities. Contrastingly, it mediates a
mild inhibitory effect on CYP2C19 and P-gp, while
imposing a pronounced inhibition on the activity of
CYP2D6 and CYP3A enzymes. The investigative
outcomes underscore a spectrum of induction and
inhibition effects, thereby enriching the
comprehension of drug interaction mechanisms
inherent to TPV/RTV. Such insights are
indispensable for the refinement of clinical
deployment strategies for TPV/RTV.
The research emphasizes the efficaciousness of a
phenotyping methodology in the prognostication of
complex drug interactions and advocates for the
application of biomarker probes in clinical
pharmacokinetics. However, the translation of these
findings to alternative therapeutic agents that display
similar mixed inhibitory and inductive propensities
should be approached with circumspection. The
solicitation of further investigative efforts is
necessary to unravel the intricate web of interactions
that TPV/RTV may engage with a diverse array of
pharmacological entities.
3 NOVEL MECHANISMS OF
INDUCTION
3.1 Induction of CYP Enzymes in Mice
In the realm of toxicological research, the elucidation
of mechanisms underlying the induction of CYP
enzymes by xenobiotic substances remains a domain
of significant scientific inquiry. Jiang et al. embarked
on an incisive exploration of the molecular
mechanisms driving the persistent induction of
CYP1A1 and CYP1A2 enzymes after 3-
methylcholanthrene (MC) exposure, a potent
carcinogenic constituent among polycyclic aromatic
hydrocarbons (PAHs) (Jiang et al., 2009). The
investigators postulated a theory suggesting that MC
catalyzes a long-standing transcriptional activation of
the CYP1A1 and CYP1A2 gene promoters, thus
inciting extended enzyme activity (Chen et al., 2017;
Jiang et al., 2009 & Gibson et al., 2002).
To substantiate their hypothesis, the researchers
designed a study utilizing both adult male wildtype
(WT) mice and genetically modified counterparts,
engineered to harbor human CYP1A1 or murine
CYP1A2 promoter sequences. The investigative
protocol entailed the administration of MC to these
models, with subsequent assessments focused on
gauging promoter-specific transcriptional activity.
The study treated mice with MC or a vehicle
control (corn oil) daily for four consecutive days. The
researchers utilized bioluminescent imaging to assess
luciferase reporter gene expression, serving as a
proxy for promoter activity, at intervals of 1, 8, 15,
and 22 days after cessation of MC treatment.
The results indicated that MC treatment
substantially increased luciferase expression driven
by both CYP1A1 and CYP1A2 promoters in the
transgenic mice. This elevated luciferase activity
persisted for up to 22 days, with a more significant
effect observed in the CYP1A1-luc mice. The MC-
induced increases in CYP1A1 and CYP1A2 activity,
as demonstrated by luciferase expression, supported
the initial hypothesis regarding sustained
transcriptional activation (Chen et al., 2017; Jiang et
al., 2009 & Gibson et al., 2002).
Endogenous CYP1A1 and CYP1A2 expression
was persistently induced in WT, CYP1A1-luc, and
CYP1A2-luc mice, corroborating the sustained
impact of MC exposure. Analysis of the findings
indicated a 15-fold increase in CYP1A1-luc
expression (Figure 2B). In contrast, the induction of
CYP1A2-luc by MC was less pronounced (Figure 2C,
D).
These data underscore the differential and
prolonged effects of MC exposure on the induction of
CYP1A1 & CYP1A2 enzymes. The utilization of
transgenic mouse models expressing luciferase
reporters driven by the CYP1A1 and CYP1A2
promoters represents a powerful tool for elucidating
the molecular mechanisms underlying persistent
enzyme induction, particularly in the context of PAH-
induced carcinogenesis.
ICREE 2024 - International Conference on Renewable Energy and Ecosystem
80
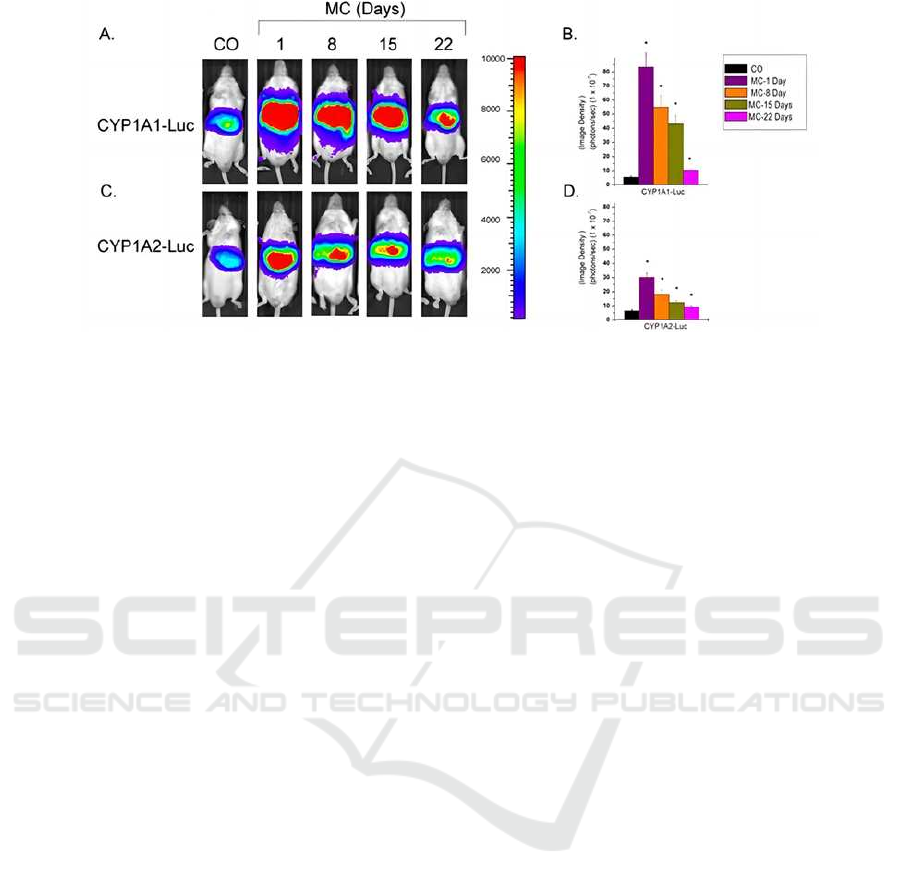
Figure 2: Bioluminescent imaging of CYP1A1-luc & CYP1A2-luc mice of MC treatment (Dumond et al., 2010).
3.2 Novel CYP Induction Mechanism
by YAP/TEAD Inhibitors in
Human Hepatocytes
DDIs which is mediated by the induction of
CYPenzymes represent a significant challenge in
drug development, necessitating comprehensive
assessment during preclinical studies. A recent study
focuses on the Hippo signalling pathway, which is
known to govern cell fate, proliferation, and
apoptosis, emphasizing its significance in the
regulation of cellular processes (Zhang et al., 2024).
At the molecular fulcrum of the Hippo signalling
cascade lies the intricate intracellular trafficking of
Yes-associated protein (YAP) and transcriptional
coactivator with PDZ-binding motif (TAZ), which
transits between the cytoplasm and nucleus.
Ordinarily, in the quiescent state of the Hippo
pathway, YAP/TAZ are permitted nuclear ingress,
instigating the transcription of genes that promulgate
cell proliferation and survival. This nuclear
localization embodies the core of a cellular growth-
promoting regime. In stark contrast, the activation of
the Hippo pathway signals for a shift—YAP/TAZ are
sequestered within the cytoplasm, an event that
curtails their role as transcriptional regulators and, by
extension, serves as a biochemical clamp on cellular
proliferation. The dichotomy of YAP/TAZ
localization thus constitutes a critical regulatory axis
in cell growth control, embodying a cellular
barometer that modulates gene expression in
accordance with the proliferative or inhibitory cues of
the Hippo pathway (Chen et al., 2017; Jiang et al.,
2009 & Gibson et al., 2002).
The YAP/TEAD (TEA domain family member)
signalling pathway has gained attention as a potential
therapeutic target in oncology due to its role in cell
growth regulation. Various inhibitors of YAP/TEAD
are progressing through clinical development, each
with distinct chemical structures and mechanisms of
action. The pathway is also influenced by cell-cell
contacts and biomechanical factors. YAP/TEAD
activity is further modulated by cell geometry, which
contributes to the complexity of the system.
Oliva-Vilarnau et al. conducted a pivotal study to
evaluate the potential for CYP induction by assessing
multiple YAP/TEAD inhibitors with varying
selectivity profiles for TEAD isoforms (Oliva-
Vilarnau et al., 2023). This study explored both
traditional 2D cultures & 3D spheroids of PHH. The
results indicated that YAP/TEAD inhibition caused
extensive CYP enzyme induction in 2D monolayers
but significantly reduced induction in 3D spheroids,
suggesting a critical relationship between cell
geometry and CYP regulation.
Further in-depth analysis through RNA
sequencing revealed that YAP/TEAD signalling was
more pronounced in 2D cultures compared to 3D,
likely due to alterations in mechanoenzyme. The
hyperactivation of YAP/TEAD in these cultures
contributed to increased activity of other interacting
transcription factors. This hyperactivation led to
hepatocyte dedifferentiation, with a corresponding
increase in hepatic function, including CYP enzyme
induction. This induction was, therefore, an indirect
consequence of YAP/TEAD inhibition.
These findings underscore the relevance of the
Hippo pathway in drug development and its broader
implications in pharmacokinetics. It highlights the
necessity for organotypic 3D cultures in preclinical
studies to better simulate clinical conditions and
accurately assess pharmacokinetic profiles.
Consequently, the results advocate for advanced
testing models to refine the drug development process
and enhance safety evaluations.
Progress in CYP Enzymes Mechanisms of Induction and Its Applications
81
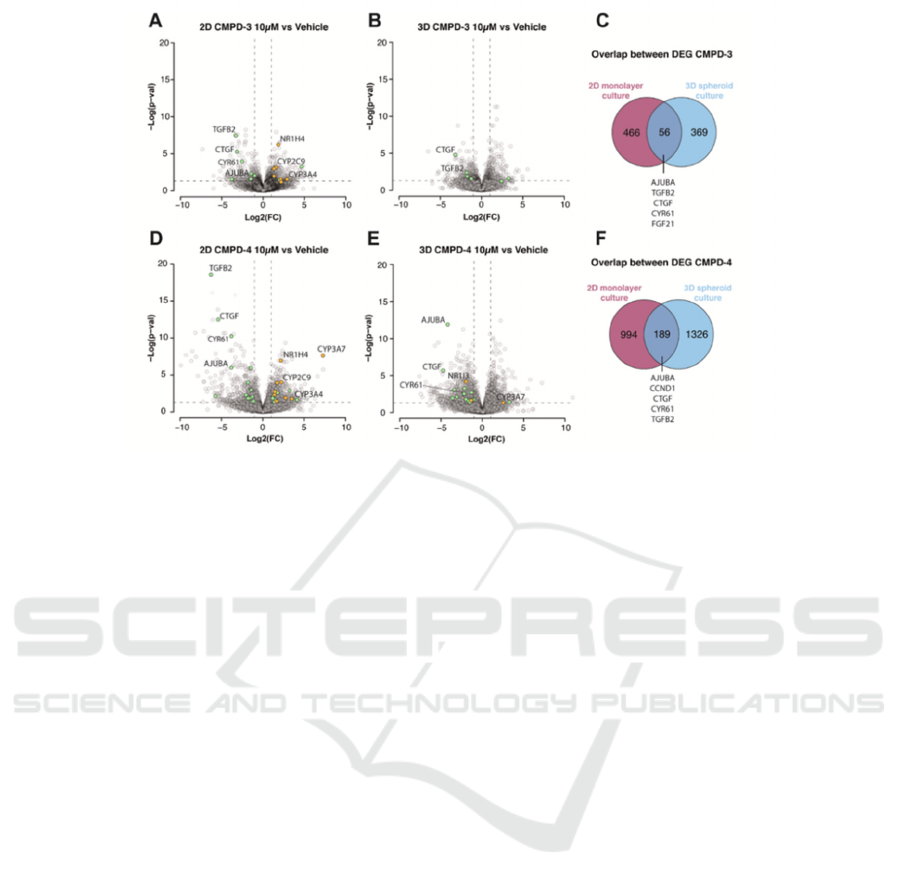
Figure 3: Comparative Gene Expression Response to YAP/TEAD Inhibition in 2D vs. 3D Primary Human Hepatocyte
Cultures (Oliva-Vilarnau et al., 2023)
A-B: The figure presents volcano plots illustrating
the gene expression impact of Compound 3 (CMPD-
3) on PHHcultured in two dimensions (2D) (A) and
three dimensions (3D) (B). Differentially expressed
genes (DEGs) are highlighted.
C: A Venn diagram details the intersection of
DEGs between 2D and 3D PHH cultures under
CMPD-3 influence, underscoring the distinctive gene
expression profiles elicited by YAP/TEAD inhibition
in differing culture formats.
D-E: Volcano plots convey the gene expression
ramifications of Compound 4 (CMPD-4) on PHH in
2D and 3D contexts. As with CMPD-3, DEGs
connected to ADME are denoted in yellow, and those
linked to Hippo signalling are in green.
F: Another Venn diagram showcases the
commonalities and discrepancies of DEGs across 2D
and 3D PHH cultures when subjected to CMPD-4,
highlighting the dependency of cellular responses to
YAP/TEAD inhibition on the cultural environment.
4 CLINICAL TRIALS
In the domain of drug metabolism research, in vitro
assays are a cornerstone; however, their predictive
validity for in vivo CYP induction is inherently
limited. Hence, in vivo assays emerge as the gold
standard, offering a more faithful reflection of CYP
induction within a living system. The direct
measurement of enzyme quantity and activity in vivo
presents practical challenges, especially in humans.
Consequently, an indirect methodology prevails,
primarily involving comparative analysis of a drug's
AUC before and after the introduction of a novel drug
entity or potential inductive agent.
Preclinical evaluation often recruits animal
models—ranging from mice and rats to monkeys and
dogs—to ascertain CYP induction as a precursor to
human testing. It is crucial to recognize the distinct
discrepancies in enzyme systems and receptor
affinities across species, which often result in
divergent metabolic responses. For example,
omeprazole's induction effect on CYP1A2 is
exclusive to humans and does not extend to murine or
rat models. To mitigate such species-specific
limitations, research has pivoted towards the
development of humanized mice through genetic
engineering or the engraftment of human hepatocytes
into immunocompromised mice, circumventing the
confounding influence of murine hepatic enzymes.
Animal models, notwithstanding the interspecies
variation in CYP induction, serve to generate initial
pharmacokinetic profiles. Nevertheless, human
subjects remain the epitome for assessing CYP
induction. In human studies, the characterization of
enzyme induction is conducted using selected CYP
probe substrates, adhering to rigorous criteria such as
enzyme specificity, minimal cross-enzyme inhibition,
and optimal pharmacokinetic characteristics, like
minimal rapid metabolism or shorter half-lives.
ICREE 2024 - International Conference on Renewable Energy and Ecosystem
82
An alternative in vivo measurement strategy
involves evaluating pharmacological parameters like
the EC50 and Emax, mindful of the interindividual
variances that arise due to CYP polymorphisms,
which can influence the response to induction probes.
Per FDA directives, data from in vitro assays and
preliminary clinical assessments should inform the
decision to advance to comprehensive human in vivo
or clinical evaluations. A drug is earmarked for in
vivo investigation only if it elicits an induction
exceeding 40%. Furthermore, the FDA's regulatory
framework allows for the exclusion of certain
enzymes from in vivo scrutiny if in vitro results are
conclusively negative (Ghosh et al., 2023).
4.1 Time Course Quantification of
CYP3A Modulation with Micro
Dosed MDZ
The enzymatic activity of CYP3A is important in the
biotransformation and clearance of various
pharmaceuticals, predominantly in hepatic and
enteric regions. Perpetrator drugs that modulate the
activity of CYP3A enzymes can profoundly influence
the pharmacokinetic profile of CYP3A substrates,
potentially culminating in clinically relevant DDIs.
Traditional studies on DDIs tend to prioritize static
exposure levels as endpoints, thus overlooking the
dynamic nature of these enzymatic interactions over
time. A comprehensive understanding of the temporal
modulation of CYP3A could shed light on critical
periods of altered enzymatic activity, thereby
enhancing the strategic planning and oversight of
DDIs in clinical settings.
To bridge this knowledge gap, Stevison, F et al.
incorporated the use of surrogate probe substrates,
such as MDZ, in their investigation of drug
interactions (Stevison et al., 2019). Given that MDZ
is extensively metabolized by CYP3A, its
pharmacokinetic profile serves as a reliable reflection
of CYP3A activity. The linearity of MDZ
pharmacokinetics across a broad dosage spectrum
facilitates the assessment of CYP3A activity without
eliciting notable pharmacodynamic responses.
This clinical investigation aimed to delineate the
temporal patterns and degree of in vivo modulation of
hepatic CYP3A activity by various perpetrator drugs.
The researchers implemented a continuous micro
dosing regimen involving intravenous MDZ, which
allowed for precise quantification of metabolic DDIs
in a healthy cohort. The study also endeavored to
characterize different modulatory mechanisms.
The study's protocol included 24 healthy
participants who received an initial bolus of
intravenous MDZ. Subjects were stratified into four
cohorts, each receiving a distinct CYP3A perpetrator
drug: voriconazole, rifampicin, or efavirenz, with two
placebo-controlled individuals per group. Following
the MDZ infusion, perpetrator drugs were introduced
after a 2-hour interval. Regular blood sampling
facilitated the measurement of MDZ and its primary
metabolite, 1'-hydroxyMDZ. The study's foremost
aim was to quantify the temporal modulation of
CYP3A activity by comparing MDZ clearance
among the treatment and placebo cohorts.
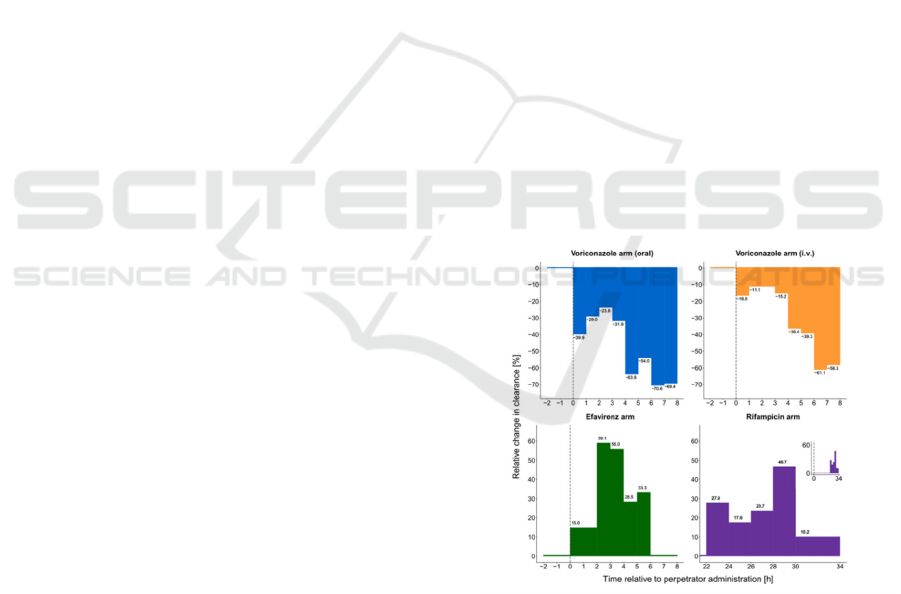
The findings demonstrated unique temporal
signatures and intensities of CYP3A modulation by
each perpetrator drug. Notably, efavirenz, recognized
as a CYP3A enhancer, displayed a swift onset of
modulation, attaining peak impact within 2 to 3 hours.
Conversely, rifampicin, a CYP3A inductor,
manifested a protracted onset, with maximal impact
noted after 28 to 30 hours, and subsequently a rapid
return to baseline within 1 to 2 hours. Voriconazole,
in both oral and intravenous forms, displayed a
sustained inhibitory effect on CYP3A, maintaining
suppression over the duration of the sampling
interval, which extended to 8 hours post-
administration. The study charted the differential
peak clearance alterations induced by efavirenz,
rifampicin, and both administrations of voriconazole,
as depicted in Figure 4.
Figure 4: The percentage change in MDZ clearance. over
time in comparison to the placebo group, following the
administration of perpetrator drugs (Stevison et al., 2019).
The perpetrator drugs are categorized into
different arms. The vertical dashed line indicates the
time when the perpetrator drug was administered.
Progress in CYP Enzymes Mechanisms of Induction and Its Applications
83

4.2 Translating Cytochrome P450
Downregulation from In Vitro to In
Vivo
Surrogate probe substrates such as MDZ are
instrumental in drug interaction research, providing a
scientifically robust method to evaluate CYP enzyme
activity. MDZ, primarily metabolized by the CYP3A
isozyme, serves as an accurate barometer for CYP3A
functionality due to its linear pharmacokinetics across
extensive dosing ranges, thus enabling the assessment
of CYP3A without eliciting marked pharmacological
outcomes (Ingelman, 2004).
A 2019 investigation by Li et al. delved into the
impact of all-trans-retinoic acid (atRA), the
metabolite of vitamin A on CYP2D6 expression in
diverse experimental systems (Li et al., 2019). Prior
studies identified that atRA diminishes CYP2D6
expression in cellular models and murine systems by
activating the transcriptional corepressor small
heterodimer partner (SHP). Yet, whether this
suppressive action translates to human physiology
remained ambiguous. Notably, atRA does not
interfere with the enzymatic function of CYP2D6,
offering a unique vantage point to study DDIs that
arise exclusively from transcriptional downregulation
(Chen et al., 2017; Jiang et al., 2009 & Gibson et al.,
2002).
The atRA isomer 13cisRA, is particularly suited
for scrutinizing the translational aspects of CYP
downregulation. Following administration, 13cisRA
is isomerized to atRA and subsequently metabolized
into 4-oxo-13cisRA in humans. Consequently, any
DDIs observed post-13cisRA administration could be
ascribed to 13cisRA itself or a collective effect of
these metabolites.
The primary objective of Li et al.'s research was to
elucidate the impact of three compounds, namely
13cisRA, atRA, and 4-oxo-13cisRA, on the
expression of CYP2D6 within hepatic cells in humans
(Li et al., 2019). Additionally, the study aimed to
determine whether the findings from in vitro
experiments could serve as predictors of potential
DDIs in clinical settings. The research findings
suggested a theoretical reduction of approximately
50% in CYP2D6 activity following the
administration of 13cisRA, as observed in in vitro
assays. However, analysis of clinical data,
specifically the area under the plasma concentration-
time curve for dextromethorphan, a substrate of
CYP2D6, revealed only a minor increase in the drug's
metabolic clearance after 13cisRA therapy. Similarly,
in murine models, the administration of 4-oxo-
13cisRA resulted in increased mRNA expression of
several Cyp2d isoforms; however, this effect did not
strongly correlate with in vivo modulation of
CYP2D6 activity.
Moreover, a modest in vitro induction of CYP3A4
in PHH was associated with a correspondingly minor
induction in vivo, thereby presenting a disparity
between in vitro observations of CYP downregulation
and the manifestation of clinical DDIs. These
observations accentuate the need for an enriched
comprehension of the mechanisms governing CYP
downregulation to refine the prediction and
management of DDIs in a clinical context.
5 CONCLUSION
The scientific quest to understand the induction of
CYP enzymes has garnered considerable focus within
the realms of drug development and toxicology
(Carroccio, 1994). The goal is to foster a deeper, more
precise scientific understanding. Although there has
been considerable progress in decoding the
mechanisms that underlie CYP induction, the
intricacies of these molecular processes demand
further elucidation. This need is heightened by the
ongoing introduction of novel pharmaceuticals and
environmental agents, each necessitating
comprehensive evaluations to assess their potential
for CYP induction and the resultant risk of
undesirable side effects.
The field has seen substantial advancements with
the introduction of innovative models for the study of
CYP induction. These models operate both within
controlled laboratory environments (in vitro) and
living organisms (in vivo), significantly bolstering
our capacity to examine and anticipate the behaviour
of CYP induction. They have markedly enriched our
knowledge of the signalling pathways and the diverse
array of factors that govern CYP induction, including
genetic variability and environmental influencers.
Nevertheless, the predictive accuracy for CYP
induction in relation to newly synthesized compounds
is still encumbered by the multifaceted nature of these
biological pathways.
Currently, the drug development industry is
equipped with well-established experimental
protocols for investigating the induction of CYP
enzymes. The data derived from these laboratory
analyses provide a foundational guide for subsequent
biological investigations and are integral to the
creation of predictive models that mirror the
pharmacokinetic processes observed in actual
physiological conditions. Despite these advanced in
silico tools, empirical studies in human populations
ICREE 2024 - International Conference on Renewable Energy and Ecosystem
84

are indispensable for a conclusive portrayal of the
effects of CYP induction and inhibition, a
requirement that is particularly salient for the
attainment of regulatory approval. It is fortuitous that
ongoing innovations in research methodologies have
sharpened the ability to detect potential drug
interactions that are mediated by CYP enzymes early
in the drug development cycle, thereby mitigating
unforeseen adverse effects in clinical settings. Early
recognition of such interactions is crucial, steering the
course of drug development away from entities that
exhibit strong inhibitory or inductive effects on CYP
enzymes. Nonetheless, we must acknowledge the
presence of yet unidentified agents, possibly present
in our diet, herbal treatments, and environmental
exposures.
The profound enhancement of our understanding
of CYP-mediated interactions has refined the drug
development paradigm and the implementation of
computational tools and databases to support
medication prescribing practices has greatly
facilitated their clinical deployment. These
advancements are particularly vital considering the
vast repository of data on DDIs, which presents a
formidable challenge for clinicians to master single-
handedly. It is pertinent to note that while the design
of drugs has traditionally cantered on improving
metabolic stability to diminish CYP-related
interactions, it is also essential to consider the
potential for interactions mediated by biological
transport mechanisms.
Although the capability to predict CYP inhibition
and induction is generally reliable, exceptional cases
continue to emerge that defy expectations. For
instance, the synergistic interaction between non-
activating compounds and the PXR, resulting in
receptor activation, exemplifies the complexity of
predicting drug interactions. These synergies may
manifest among pharmaceutical agents or in
scenarios of exposure to complex environmental
mixtures, relevant in toxicology. Hence, despite the
substantial body of knowledge regarding CYP
inhibition and induction accrued over the years,
ongoing research is imperative. The landscape of
drug interaction remains dynamic, with the ever-
present prospect of unearthing new insights.
REFERENCES
Pelkonen, O.; Turpeinen, M.; Hakkola, J.; Honkakoski, P.;
Hukkanen, J.; Raunio, H. (2008). Inhibition and
induction of human cytochrome P450 enzymes:
Current status. Arch. Toxicol. 82, 667–715.
Li Y, Meng Q, Yang M, Liu D, Hou X, Tang L, Wang X,
Lyu Y, Chen X, Liu K, Yu AM. (2019). Current trends
in drug metabolism and pharmacokinetics. Acta
Pharmaceutica Sinica B. Nov 1;9(6):1113-44.
Lin, J. H. (2006). CYP induction mediated drug
interactions: Invitro assessment and clinical
implications. Pharm. Res. 23,1089–1116.
Schuetz, J.D.; Schuetz, E.G.; Thottassery, J.V.; Guzelian,
P.S.; Strom, S.; Sun, D. (1996). Identification of a novel
dexamethasone responsive enhancer in the human
CYP3A5 gene and its activation in human and rat liver
cells. Mol. Pharmacol. 49, 63–72.
Pascussi,J.M.;Drocourt,L.;Gerbal Chaloin, S. ; Fabre, J.
M. ; Maurel, P. ; Vilarem, M. J. Dual effect of
dexamethasoneon CYP3A4 gene expression in human
hepatocytes. (2001). Sequential role of glucocorticoid
receptor and pregnane X receptor. Eur. J. Biochem. 268,
6346–6358.
Chen, Y.; Zeng, L.; Wang, Y.; Tolleson, W.H.; Knox, B.;
Chen, S.; Ren, Z.; Guo, L.; Mei, N.; Qian, F.; etal.
(2017). The expression, induction and pharmacological
activity of CYP1A2 are post-transcriptionally regulated
by microRNA hsa-miR-132-5p. Biochem. Pharmacol.
145, 178–191.
Bernasconi, C., Pelkonen, O., Andersson, T.B., Strickland,
J., Wilk-Zasadna, I., Asturiol, D., Cole, T., Liska, R.,
Worth, A., Müller-Vieira, U. and Richert, L. (2019).
Validation of in vitro methods for human cytochrome
P450 enzyme induction: Outcome of a multi-laboratory
study. Toxicology in Vitro, 60, pp.212-228.
Dumond, J.B., Vourvahis, M., Rezk, N.L., Patterson, K.B.,
Tien, H.C., White, N., Jennings, S.H., Choi, S.O., Li, J.,
Wagner, M.J. and La‐Beck, N.M. (2010). A Phenotype–
Genotype Approach to Predicting CYP450 and P‐
Glycoprotein Drug Interactions with the Mixed
Inhibitor/Inducer Tipranavir/Ritonavir. Clinical
Pharmacology & Therapeutics, 87(6), pp.735-742.
Jiang, W., Wang, L., Zhang, W., Coffee, R., Fazili, I.S. and
Moorthy, B. (2009). Persistent induction of cytochrome
P450 (CYP) 1A enzymes by 3-methylcholanthrene in
vivo in mice is mediated by sustained transcriptional
activation of the corresponding promoters. Biochemical
and biophysical research communications, 390(4),
pp.1419-1424.
Stevison F, Kosaka M, Kenny JR, et al. (2019). Does In
Vitro Cytochrome P450 Downregulation Translate to In
Vivo Drug-Drug Interactions? Preclinical and Clinical
Studies With 13-cis-Retinoic Acid. Clin Transl Sci.
12(4):350-360.
Ghosh R, Nayan MI, Mitu MM, Nandi T. (2023). Common
Approaches of Cytochrome P450 (CYP) Induction
Assays. International Blood Research & Reviews. Jan
18;14(1):6-14.
Oliva-Vilarnau, N., Vorrink, S.U., Büttner, F.A., Heinrich,
T., Sensbach, J., Koscielski, I., Wienke, D., Petersson,
C., Perrin, D. and Lauschke, V.M. (2023). Comparative
analysis of YAP/TEAD inhibitors in 2D and 3D cultures
of primary human hepatocytes reveals a novel non-
canonical mechanism of CYP induction. Biochemical
Pharmacology, 215, p.115755.
Progress in CYP Enzymes Mechanisms of Induction and Its Applications
85

Nassar, Y.M., Hohmann, N., Michelet, R., Gottwalt, K.,
Meid, A.D., Burhenne, J., Huisinga, W., Haefeli, W.E.,
Mikus, G. and Kloft, C. (2022). Quantification of the
time course of CYP3A inhibition, activation, and
induction using a population pharmacokinetic model of
microdosed midazolam continuous infusion. Clinical
Pharmacokinetics, 61(11), pp.1595-1607.
Gibson GG, Plant NJ, Swales KE, Ayrton A, El-Sankary W.
(2002). Receptor-dependent transcriptional activation
of cytochrome P4503A genes: induction mechanisms,
species differences and interindividual variation in
man. Xenobiotica. Jan 1;32(3):165-206.
Zhang R, Wang XX, Xie JF, Yao TT, Guo QW, Wang Q,
Ding Z, Zhang JP, Zhang MR, Xu LC. (2024).
Cypermethrin induces Sertoli cell apoptosis through
endoplasmic reticulum-mitochondrial coupling
involving IP3R1-GRP75-VDAC1. Reproductive
Toxicology. Jan 29:108552.
Si C, Yang H, Wang X, Wang Q, Feng M, Li H, Feng Y,
Zhao J, Liao Y. (2024). Toxic effect and mechanism of
β-cypermethrin and its chiral isomers on HTR-8/SVneo
cells. Pesticide Biochemistry and Physiology. May 1;
201:10584.
Carroccio, A.; Wu, D.; Cederbaum, A.I. (1994) Ethanol
increases content and activity of human cytochrome
P450 2E1 in a transduced HepG2 cell line. Biochem.
Biophys. Res. Commun. 203, 727–733.
Ingelman-Sundberg, (2004). M. Human drug metabolising
cytochrome P450 enzymes: properties and
polymorphisms. Naunyn Schmiedebergs Arch.
Pharmacol. 369, 89–104.
ICREE 2024 - International Conference on Renewable Energy and Ecosystem
86
