
Neural Population Decoding and Imbalanced Multi-Omic Datasets
for Cancer Subtype Diagnosis
Charles Theodore Kent
1
, Leila Bagheriye
2
and Johan Kwisthout
2
1
School of Artificial Intelligence, Radboud Universiteit, Houtlaan 4, Nijmegen, Netherlands
2
Donders Institute for Brain, Cognition & Behaviour, Radboud Universiteit, Houtlaan 4, Nijmegen, Netherlands
Keywords: Cancer Diagnosis, Multi-Omics, Population Decoding, Spiking Neural Networks, Winner-Take-All,
Hierarchical Bayesian Network, Self-Organising Maps.
Abstract: Recent strides in the field of neural computation has seen the adoption of Winner-Take-All (WTA) circuits to
facilitate the unification of hierarchical Bayesian inference and spiking neural networks as a neurobiologically
plausible model of information processing. Current research commonly validates the performance of these
networks via classification tasks, particularly of the MNIST dataset. However, researchers have not yet
reached consensus about how best to translate the stochastic responses from these networks into discrete
decisions, a process known as population decoding. Despite being an often underexamined part of SNNs, in
this work we show that population decoding has a significant impact on the classification performance of
WTA networks. For this purpose, we apply a WTA network to the problem of cancer subtype diagnosis from
multi-omic data, using datasets from The Cancer Genome Atlas (TCGA). In doing so we utilise a novel
implementation of gene similarity networks, a feature encoding technique based on Kohoen’s self-organising
map algorithm. We further show that the impact of selecting certain population decoding methods is amplified
when facing imbalanced datasets.
1 INTRODUCTION
Multi-omics data integration in cancer diagnosis
refers to the integration of information from various
biological "omics" e.g., genomics, transcriptomics,
metabolomics, to provide a more comprehensive
understanding of the molecular landscape of cancer.
Spiking neural networks (SNNs) are a
neurobiologically inspired method of information
processing which aim to solve tasks using plausible
models of neuron dynamics (Yamazaki et al., 2022).
Much like in biological brains, neurons in SNNs are
linked through excitatory and inhibitory connections,
and propagate information via discrete electrical
signals known as spikes (Yamazaki et al., 2022;
Himst et al., 2023). An important feature of SNNs is
that their activations are stochastic (Ma & Pouget,
2009), and so presenting a network with the same
stimulus multiple times will likely result in varying
responses. We can gain more insight into the network
through sampling the distribution of responses when
presenting a stimulus over multiple time steps,
simulating exposure for a given length of ‘biological
time’ (Guo et al., 2017). The responses of the network
during this window can be quantified by counting the
number of times each neuron spikes, referred to as a
spike count code (Grün & Rotter, 2010).
Alternatively, some research focuses on the time-
dependent relationship of spiking neurons, for
instance by weighting neuron responses more highly
based on how quickly they fire (Grün & Rotter, 2010;
Shamir, 2009; Beck et al., 2008).
In order to extract information from SNNs, we
examine the spikes generated by a population of
neurons in response to a stimulus. The process of
presenting a stimulus to the network to generate these
spikes is known as population encoding, and
conversely the process of obtaining estimates from
the neuron activity patterns is known as population
decoding (Ma & Pouget, 2009). Together, these two
opposite processes are referred to as population
coding. Population coding can be used in conjunction
with SNNs to gain practical insights into how a
system of spiking neurons tackles the task of learning
(Ma & Pouget, 2009).
Approaching the question from a more theoretical
standpoint, Bayesian inference is hypothesised to be
a key component of information processing within the
Kent, C., Bagheriye, L. and Kwisthout, J.
Neural Population Decoding and Imbalanced Multi-Omic Datasets for Cancer Subtype Diagnosis.
DOI: 10.5220/0012454200003657
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 17th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2024) - Volume 1, pages 391-403
ISBN: 978-989-758-688-0; ISSN: 2184-4305
Proceedings Copyright © 2024 by SCITEPRESS – Science and Technology Publications, Lda.
391

brain (Guo et al., 2017), including in areas of
cognition and decision making (Shamir, 2009).
Handling uncertainty when understanding their
environment is critical to the survival of many
organisms, and Bayes’ theorem provides a
biologically plausible framework for the brain’s
probabilistic nature (Kersten et al., 2004). Based on
electrophysiological recordings, neurons appear to
process information in a hierarchical manner, which
can similarly be modelled as hierarchical Bayesian
inference (Lee & Mumford, 2003).
Until recently, research into SNNs and hierarchical
Bayesian models of the brain have remained separated
by the computational complexity of performing exact
inference (Guo et al., 2017). To overcome this
problem, the variational principle can be employed to
decompose the difficult exact inference into an
optimisation problem which is easier to solve (Guo et
al., 2017; Friston, 2010). Following this approach,
spiking neural networks have been able to implement
hierarchical Bayesian models through use of neural
circuits such as the Winner-Take-All (WTA) circuit
(Guo et al., 2017; Nessler et al., 2013). In a WTA
circuit, a layer of excitatory neurons is linked to a
corresponding layer of inhibitory neurons. Whenever
an excitatory neuron fires, an inhibitory signal is
generated in response which resets the neuron
membrane potentials to baseline and updating the
weights of each connection (Himst et al., 2023). When
coupled with the spike-timing dependent plasticity
(STDP) learning rule, this framework enables neurons
to learn structural representations from stimuli in an
unsupervised manner (Himst et al., 2023; Guo et al.,
2017; Nessler et al., 2013).
Utilising this technique, experimental research into
hierarchical Bayesian WTA networks have begun
reporting results on the benchmark dataset MNIST
(Himst et al., 2023; Guo et al., 2017; Diehl & Cook,
2015; Nessler et al., 2013; Querlioz et al., 2013).
Generally, the accuracy of these models on the
MNIST test set is in the range of 80-85%, with some
works (Guo et al., 2017; Diehl & Cook, 2015)
achieving accuracies as high as 95% with an
optimised set of model hyperparameters. Whilst the
reported results are promising and show the potential
applications of WTA networks for real-world
problems, current research shows little consideration
to the significance of population coding in
classification tasks.
One point of contention revolves around the choice
of population decoding method used to turn neuronal
responses into a discrete prediction for classification.
In the vast majority of experiments (Himst et al.,
2023; Guo et al., 2017; Diehl & Cook, 2015; Querlioz
et al., 2013), neurons are assigned to the class for
which they spike most frequently over a given dataset
in an a posteriori fashion. Typically, the responses
used to make this assignment are collected by
presenting the network samples from a training set
(Diehl & Cook, 2015; Nessler et al., 2013; Querlioz
et al., 2013), however in some cases (Himst et al.,
2023; Guo et al., 2017) this step is performed over the
test set instead. Unfortunately, the combination of a
posteriori assignment and utilisation of test set labels
can be shown to lead to high degrees of bias, which
we elaborate on in Section 4.
Beyond data subset selection, there are
discrepancies between population decoding practices
adopted by researchers. By far the most common
approach to population decoding (Himst et al., 2023;
Guo et al., 2017; Diehl & Cook, 2015; Nessler et al.,
2013; Querlioz et al., 2013; Ma & Pouget, 2009) is to
assign each neuron a single label based on the class
of stimulus for which the neuron responds most
highly. Then, upon presentation of a test stimulus, the
responses of each neuron are averaged per class,
before selecting the class with the highest average
firing rate. We term this methodology the class
averaging decoder, and give a full mathematical
description in Section 2.
In Nessler et al. (2013), the population decoding
step is hand-performed by a human supervisor by
examining the weights of the trained model. Whilst
this approach is somewhat reasonable in the context
of MNIST, where it is relatively simple for a human
to discern the correct classification by eye, it clearly
leaves a lot to be desired. Firstly, the process is not
scalable, as some notable experimental results (Guo
et al., 2017; Diehl & Cook, 2015) recommend using
many thousands of output neurons for optimal
classification performance. Moreover, not all neurons
in the output population will learn a representation
that is easily recognisable. A given neuron may be
tuned to detect certain sub-features within the image,
be half-way between two distinct classes, or fail to
learn a meaningful representation entirely and have
weights resembling random noise or arbitrary blobs
(Himst et al., 2023; Nessler et al., 2013; Querlioz et
al., 2013). In these cases, human bias can easily creep
into the prediction process, and so a more
mathematically grounded approach is desirable.
Querlioz et al. (2013) use a validation subset of
1,000 “well-identified” images to form their neuron-
class associations. Querlioz et al. (2013) also point
out that the labelling process need not occur
concurrently with training, but can be done at a later
stage. Furthermore, Querlioz et al. (2013) suggests an
avenue for future work could be the coupling of an
BIOINFORMATICS 2024 - 15th International Conference on Bioinformatics Models, Methods and Algorithms
392

SNN to a supervised network to perform the
population decoding step, a concept that we will be
investigating further in this paper.
Notably, in all of the prior discussed approaches,
the population decoding step is treated as separate
from the SNN model. However, it could be argued
that this step must occur somewhere within the brain,
as we are ultimately able to resolve sources of
uncertainty down into concrete choices. The meta-
task of mapping responses from an arbitrarily large
population of neurons down to a single discrete
decision is generally not approached from a
biologically plausible perspective (Ma & Pouget,
2009). As discussed, most methodologies (Himst et
al., 2023; Guo et al., 2017; Diehl & Cook, 2015;
Querlioz et al., 2013) use a running total of the
neuronal responses over each stimulus presented to
the network to determine each neuron’s class. Yet, it
seems implausible for brains to store and update a
counter of every time they have seen a certain class
of stimuli throughout their whole lives, and reference
that counter to make decisions.
A possible alternative to this methodology could be
to incorporate a supervised model to perform the
population decoding step. For instance, a multivariate
logistic regression model requires only the use of a
weight, bias and sigmoid activation function;
components which have each independently been
shown to be neurobiologically plausible (Hao et al.,
2020). Another benefit of logistic regression is that the
model can be trained in an online fashion, updating the
weights and bias upon presentation of each individual
stimulus, and thereby avoiding the necessity of
viewing the entire dataset simultaneously. Whilst this
supervised approach is strictly not biologically
plausible, as the learning and inference steps are not
spike-based (Hao et al., 2020), these factors at least
bring us closer to the desired goal of a complete model
of neural information processing.
One of the key factors to be considered in the
practical application of Bayesian WTA networks is
the role of class imbalance. We posit that the
approaches to population decoding which we have
discussed play a sizeable role in the system’s overall
ability to perform classification, and that class
imbalance has a strong impact on said performance.
Contemporary research primarily focuses on the
MNIST dataset, which has equally balanced class
samples by default, and so issues which arise in this
area have yet to be elucidated. If research is to move
beyond the benchmark domain, handling class
imbalance is a necessity, as innumerable real-world
problems possess this property.
The purpose of this research is to apply an SNN-
based hierarchical Bayesian WTA network to a non-
benchmark dataset, in order to gain further insight into
the implications of selecting various population
decoding methods. The remainder of this paper is
structured as follows. In Section 2, we introduce the
theoretical foundations of population coding in the
context of spiking neural networks, as well as the
definitions for the population decoding strategies we
will test experimentally. In Section 3, the methodology
of the experiments is described in detail. Section 4
contains the results of the aforementioned experiments,
as well as discussions of the insights gained by
practical application of these techniques. Finally,
Section 5 contains concluding remarks, suggested
areas for further research, and provides our
recommendations for future practitioners.
2 POPULATION DECODING
In this section, we provide definitions for the methods
of population decoding which will undergo
experimental evaluation in Section 4. We largely
follow the nomenclature provided by Grün & Rotter
(2010), in which they discuss resolving the ambiguity
of single-trial neuronal responses via population
coding.
Consider an experiment in which a spiking neural
network is presented with a stimulus s from a stimulus
set S. Each stimulus has one associated numeric class
label y
{0,…,C}, where C is the number of possible
classes in y, and s
y
is the classification for a given
stimulus. The spikes generated by a population of N
neurons in response to presenting a stimulus for a
fixed window of time is recorded. The neural
population response in this time period is quantified
as a vector r = r
1
,...,r
N
with dimensionality N, where
r
n
is the response of neuron n on a given trial. In this
case we are interested in spike counts, so r
n
would
therefore be the number of spikes emitted by neuron
n during the trial in the response window. With this
definition of a neural population response, we can
now perform various population decoding methods
with the response array r to associate the response
from a given stimulus s with a predicted classification
label ŷ.
A common strategy (Himst et al., 2023; Guo et al.,
2017; Diehl & Cook, 2015; Nessler et al., 2013;
Querlioz et al., 2013; Ma & Pouget, 2009) for
population decoding is to first associate each output
neuron n with a class label present in ŷ. This
association is created based on the relative strength of
neuronal responses when reacting to stimuli of each
Neural Population Decoding and Imbalanced Multi-Omic Datasets for Cancer Subtype Diagnosis
393

class within the dataset. For each stimulus s presented
to the network, we sum the spike counts r of the
output neurons inside a multi-dimensional array M
ℤ
N x C
such that
(1)
where the sums of spike count responses for each
neuron n are split by class along the c dimension.
Each element M
nc
thus corresponds to the total
amount of times a given neuron spiked for a given
class over the entire stimulus set S.
For each neuron, we can then identify the class
which has the highest spike count over the stimulus
set. In this way, we can experimentally determine a
neuron’s preferred class. We represent this associative
relationship using the vector Z = Z
1
,...,Z
N
, with
dimensionality N, defined as:
(2)
such that each element Z
n
represents the preferred
class of the corresponding neuron n. For ease of
notation, we can further treat the vector Z akin to a
function, which accepts a parameter n {1,…, N}
representing the index of a neuron as input, and
returns the preferred class of the neuron at that index.
We denote the neuron’s preferred class as ŷ
n.
(3)
From the definition presented in equations 1 & 2,
we can already see there is an implicit assumption that
the stimulus set used to construct Z contains a balanced
number of examples for each class. This is because the
sum of the spike counts is directly proportional to the
amount of times a stimulus of that class is presented to
the network. In datasets with a high degree of class
imbalance, this leads to undesirable behaviour. For
instance, a given neuron may have a far stronger
response to stimuli of one class relative to another - yet
if presented with an overwhelming number of
examples of the “less-prefferred” class, the sum of
spikes for the less-prefferred class will eventually
exceed that of the class with the higher relative spike
response rate. This leads to situations where a neuron
will be assigned a label in Z which is counter to its
observed experimental behaviour. Additional steps
must therefore be taken to rectify this behaviour if we
wish for SNNs to be performant in class-imbalanced
domains.
In the following subsections, we detail the specific
population decoding methods being evaluated in this
research. Additionally, we make note of each
method’s potential robustness to class imbalance as a
natural result of their mathematical construction.
2.1 Winner-Take-All Decoder
For this straightforward population decoding
approach, we designate the neuron with the highest
spike count response the as ‘winner’, then find its
corresponding preferred class in Z to make the final
prediction.
(4)
Overall, the simplicity of this methodology does
have significant drawbacks, as each trial is highly
sensitive to variability, and the information from the
responses of all neurons but the most active is
discarded (Ma & Pouget, 2009). In principle, it also
has little resistance to class imbalance, as an unequal
ratio of neuron labels in Z would cause a
disproportionate increase in the likelihood of the
majority class being selected.
2.2 Population Vector Decoder
Another method is to take a sum of the responses per
class, then take the class with the highest amount of
‘votes’ as the network’s prediction (Ma & Pouget,
2009). We split the responses by class in accordance
with the observed preferred class of each neuron Z
n
,
such that:
(5)
This approach is equivalent to the weighted
average shown in (Ma & Pouget, 2009), or is
sometimes referred to as ‘pooling’ the responses of
the neuronal population (Grün & Rotter, 2010). This
is also the approach implemented in Himst et al.
(2023) to achieve their results on the MNIST dataset.
Unfortunately, the population vector decoder is
greatly susceptible to class imbalance, as it is only
concerned with the class-wise sums of responses –
thus incurring the imbalance related problems which
have been discussed above in regards to construction
of the assignment vector Z.
2.3 Class Averaging
In this approach, we take the highest average firing
rate of the neurons per class to determine the
prediction. The sum of spike counts for neurons of
each class is divided by the number of neurons
assigned to that class. Formally,
BIOINFORMATICS 2024 - 15th International Conference on Bioinformatics Models, Methods and Algorithms
394

(6)
where Zc is the number of neurons assigned to class
c in the preffered class vector Z.
This is the methodology adopted by Guo et al.
(2017) and Diehl & Cook (2015), and has seen strong
experimental results when applied to the MNIST
dataset. An interesting mechanism at play in this
approach is that, in practice, the distribution of
neurons assigned to each class is proportional to the
class ratio of the stimulus dataset; a property which
we investigate further in our experimental results
Section 4. Due to this property, the class averaging
decoder is inherently more robust to class imbalance
than either of the prior discussed methods.
2.4 Firing Average
Here, we propose a novel method of population
decoding based upon the average firing rate of each
neuron. By subtracting the average firing rate from
the spike counts in the response vector, we can
thereby pay particular attention to neurons which are
abnormally highly active compared to their typical
behaviour. We first compute the vector F = F
1
,...,F
N
,
pertaining to the average firing rate of each neuron
over the a stimulus set of training data:
(7)
where |S| is the cardinality of the stimulus set S. We
can subsequently subtract the neuron-wise average to
obtain the final class estimate of the network.
(8)
This approach is theoretically beneficial in reducing
the impact of ‘over-active’ neurons, which are prone to
firing regardless of the class of the presented stimulus
– effectively acting as a regularization technique.
However, what effect this will have on handling class
imbalance is as yet unknown. Computationally
speaking, calculating the firing average does require an
additional pass over the dataset to calculate the F
vector. Also, utilizing the average spike counts means
the values of the vector F are continuous rather than
discrete, which further distances this methodology
from biological plausibility.
2.5 Logistic Regression
As suggested by Querlioz et al. (2013), a viable
approach to population decoding could be to couple
the SNN to a supervised classification model. To
demonstrate this, we consider a multivariate logistic
regression model to map network responses to
predictions. A variety of other supervised methods
could equally apply here, but as the experimental
section of this research focuses on a case with a
binary target variable, we consider the choice of
logistic regression apt for our purposes. Prediction of
the target from the network response using the trained
logistic regression model is calculated as follows:
(9)
where w is the weight vector and b is the bias term.
The training procedure is performed in an online
manner, updating the weights and bias parameters
upon each presentation of a stimulus to the SNN,
rather than over the entire dataset at once after the
SNN training procedure is completed as with the
other population decoding methods.
In regards to the imbalance problem, logistic
regression is reasonably adept at handling skewed
class ratios. In Section 3.1, we apply a logistic
regression model to a heavily imbalanced dataset and
observe strong classification performance (shown in
Figure 1). This result demonstrates the efficacy of the
technique over the original dataset, which suggests it
should likewise be able to handle imbalance in the
role of a population decoder. Additionally,
implementing a supervised model for population
decoding negates the necessity of assigning each
neuron a discrete class. We therefore do not require
the assignment vector Z as in the other described
methods, avoiding the implicit problems with class
imbalance as discussed prior.
3 METHODOLOGY
The dataset we have chosen for practically applying
hierarchical Bayesian WTA networks is from The
Cancer Genome Atlas (TCGA) (Weinstein et al.,
2013). We select the datasets concerning the diagnosis
of breast cancer (BRCA) and kidney renal clear cell
carcinoma (KIRC). The dataset is comprised of multi-
omic features relating to individual patients, including
genomics, methylation and mitochondrial RNA
sequences. Each patient has a corresponding binary
target variable which indicates their cancer diagnosis
status, either positive or negative. Importantly for this
research, there are far fewer examples of positive
diagnoses in both datasets as compared to negative
examples, allowing us to investigate the impact of class
imbalance. Furthermore, the real-world implications of
Neural Population Decoding and Imbalanced Multi-Omic Datasets for Cancer Subtype Diagnosis
395
a false positive versus false negative diagnosis are
worth considering. Patients who receive a false
positive will likely undergo further tests and ultimately
rule out the disease, whereas a false negative could
result in the patient going undiagnosed entirely, which
can have serious ramifications for treatment outcomes.
Therefore, close attention is paid to class-wise
predictive performance throughout the methodological
process.
3.1 Multi-Omic Data
In order to incorporate information from all of the
omic types present in the TCGA dataset, the files for
methylation, genomics and mitochondrial RNA were
combined into a single dataset, with each row
representing one patient mapped to approximately
80,000 omic feature columns. We perform separate
identical processes for the BRCA and KIRC cancer
subtypes. Due to their time-dependent nature, spiking
neural networks generally have a high computational
complexity. Therefore, it is imperative we perform
dimensionality reduction steps upon the dataset in
order to maintain a tractable training regime. In this
vein, we take after the approach of Fatima & Rueda
(2020) and first perform a variance threshold filter
over the data. Any feature with a variance of less than
0.2% is removed. This removes any features with zero
values recorded for more than 80% of samples,
bringing the feature countdown to approximately
20,000 for each cancer subtype.
Feature selection is the next step. There are
numerous possible algorithms which would be
appropriate to apply here; Ang et al. (2015) provides a
rich overview of available methods in the specific
context of genomic feature selection. As we have labels
for our samples, we opt to use supervised feature
selection techniques to best make use of all available
information in the dataset. In particular, the technique
of Minimum Redundancy Maximum Relevancy
(mRMR) (Ding & Peng, 2005) has been selected for
the purposes of this research. mRMR is concerned with
two metrics for feature evaluation; relevancy is a
measure of the mutual information between a feature
and the target, and redundancy measures the mutual
information between features to select mutually
maximally dissimilar genes (Ding & Peng, 2005).
These two scores are then considered with equal
weight to determine the optimal feature subset.
Using mRMR, we calculate the relevancy and
redundancy for each multi-omic feature, and start by
selecting the top 20 scoring features. We then perform
an ablation analysis upon the selected feature set by
training a logistic regression model and sequentially
eliminating the lowest scoring remaining feature,
noting the degradation in predictive performance
each time. In this case we measure performance via
F1-score, a decision which is further explained in
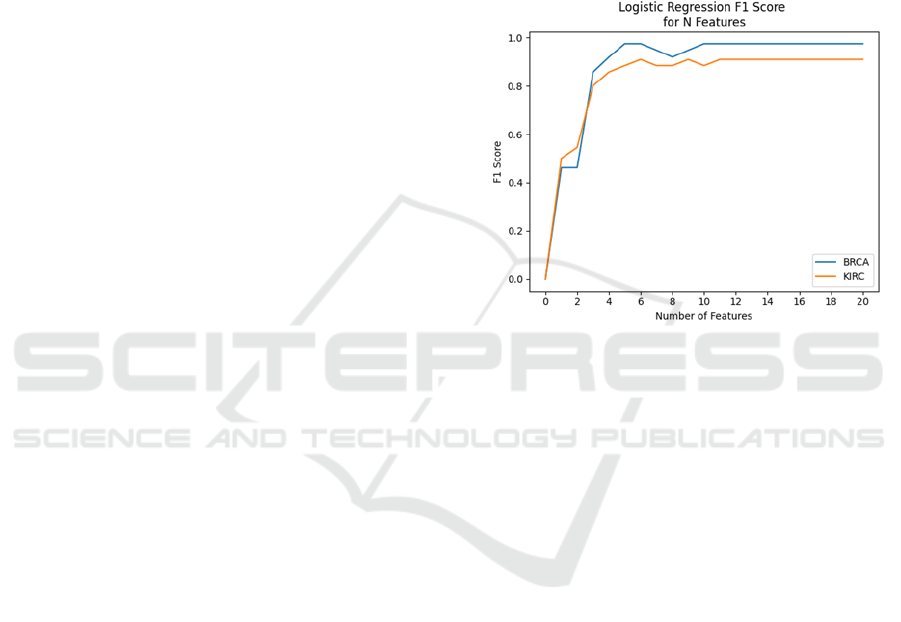
Section 4. The results of this analysis are presented in
Figure 1. Based on these results, we can see that
prediction scores reach their maximum by the
inclusion of the 10 most relevant features for BRCA
and 11 for KIRC. We therefore choose to select the
top 11 features for both cancer subtypes, so that the
pre-processing phase remains identical in either case.
Figure 1: Results of k-folds cross validation for logistic
regression models trained on a range of feature subsets.
Each time the number of features increases, the new feature
being added is the next most important by mRMR score.
3.2 Self-Organising Maps & Gene
Similarity Networks
Bayesian WTA networks of similar design to that
described in this research originate from modelling
the processing systems in the visual cortex. The
design of Bayesian WTA networks typically
incorporates sampling from a Poisson distribution
over a given frame of biological time, as a
representation of the spiking activity of upstream
neuronal firing (Himst et al., 2023; Guo et al., 2017;
Nessler et al., 2013). Therefore, an effective way to
encode information for processing in Bayesian WTA
networks is as a series of binarized images - a 2D grid
where each pixel takes the value of either 0 or 1,
varying across a time dimension. To accomplish this,
the binary images are subsequently encoded into
spike trains (Yamazaki et al., 2022), the process of
which is further described below in Subsection 3.3.
The chosen method for encoding the selected
multi-omic features into an image format is a Self-
Organising Map (SOM) (Kohonen, 1990). This
technique has been applied to TCGA cancer subtype
BIOINFORMATICS 2024 - 15th International Conference on Bioinformatics Models, Methods and Algorithms
396

diagnosis datasets in Fatima & Rueda (2020) with
marked success, and so has been selected for the
purposes of this research. A further aspect of
relevance for this technique is that it has been posited
as a biologically-plausible model of neuron self-
organisation (Kohonen, 1990). As biological
plausibility is likewise a key concern of both
hierarchical Bayesian networks and SNNs in general,
utilising this technique to encode our input data
before presenting it to the network means we can
extend this property to encompass the preprocessing
stage as well.
We employ Kohoen’s Self-Organising Map
algorithm (Kohonen, 1990) to translate each feature
to a node 2D space, where the Euclidean spatial
relationship between nodes encode semantic
information about the input data. The organisation is
done in an unsupervised manner by iteratively
computing the “best matching cell” (Kohonen, 1990)
in accordance with the distance between nodes within
topological neighbourhoods. Each node has an
associated weight vector which is updated
concurrently with its local subset, mimicking lateral
feedback connections in biophysical network models.
The algorithm will run for a set number of epochs or
until a desired convergence threshold is reached.
Upon completion, the trained SOM returns positional
coordinates for each feature in the dataset.
With our newly created spatial feature mappings,
we must now generate images representing the omic
information of each patient, known as a ‘Gene
Similarity Network’ (GSN) (Fatima & Rueda, 2020).
In Fatima & Rueda (2020), samples are encoded via an
RGB colour scheme, with each colour channel relating
to one of the three types of multi-omic data available
in the TCGA datasets. However, since our Bayesian
WTA network requires spike trains generated from
binarized images as input, using colour to encode
information is impossible in this case.
Therefore, we propose a novel method of encoding
information into the GSN by scaling and rotating each
node in accordance with the strength of feature
expression. Each feature is first normalised by Z-
score to reduce the impact of outliers. Then, to
determine the size of the GSN nodes, each feature is
scaled between a range of minimum to maximum
desired pixel sizes, proportional to the overall size of
the generated image. To determine the orientation of
each node, we similarly scale the feature columns
between the range of 0 and 180 degrees, as we opted
to use a diamond shape for each node with an order
of rotational symmetry of 2. We run the SOM
algorithm on our dataset for 5 epochs with a learning
rate of 0.05. An example of the completed GSNs is
shown in Figure 2.
Encoding information via orientation for WTA
networks is a well-studied approach given the origins
of this research focus on processing in the visual
cortex of various animal species (Grün & Rotter,
2010; Ma & Pouget, 2009). On the whole however,
binary images are a somewhat limiting format for
encoding information, as there is only one degree of
granularity for each pixel feature. This makes the task
of encoding continuous data into binary pixels a
challenging one, and we identify this as an area for
potential future research. The GSN implementation
presented here attempts to overcome this information
bottleneck by using conjointly utilising location,
rotation, and size of shapes within the image.
Figure 2: Gene Similarity Network for two patients. Each
diamond represents one feature and shares positions across
patients, but varies in size and orientation based on each
patient’s level of feature expression.
3.3 Spike Trains
The time-dependent nature of SNNs requires that
stimuli be presented to the network over an extended
period of time, so as to model biological processing
(Guo et al., 2017). However, research has shown
(Guo et al., 2021) that presenting one static input for
the duration of the presentation is both inefficient for
learning and questionable in terms of biological
plausibility. Instead, it is preferable that the stimulus
has variability over time. To accomplish this, we
follow the procedure of (Himst et al., 2023; Guo et
al., 2017; Nessler et al., 2013). The binary GSN
images are converted into Poisson spike trains, where
pixel values for each timestep are drawn from a
Poisson distribution modulated by the colour (white
or black) of that pixel in the original image. We select
a firing rate of 200hz for generating the spike trains,
and present them to the WTA network for 150ms of
simulated biological time.
Neural Population Decoding and Imbalanced Multi-Omic Datasets for Cancer Subtype Diagnosis
397
3.4 Synthetic Minority Oversampling
As certain methods of population coding are
potentially highly sensitive to class imbalance, one
particularly useful tool in this circumstance is that of
Synthetic Minority Oversampling Techniques
(SMOTE) (Chawla et al., 2002). SMOTE offers an
effective way to mitigate imbalance-related issues by
including additional synthetic examples of the
minority class in the training set. Although there are
numerous potential methods to generate new
synthetic datapoints, for the purposes of this research
we deem it sufficient to simply over-sample the
minority class up to a desired ratio of class imbalance.
This is due to the fact that several of the population
decoding methodologies described in Section 2 are
heavily affected by class imbalance; in these cases,
the impact of training on more varied samples has a
negligible impact on predictions as compared to
merely re-balancing the training class distribution.
We define the class ratio α of a set as:
(10)
where C
m
is the number of samples in the minority
class, and C
M
is the number of samples in the majority
class (Imbalanced-learn, 2016). Prior to resampling,
the BRCA dataset has a ratio of α=0.066 and KIRC
has a ratio of α=0.091. We investigate the impact of
various α ratios experimentally in Section 4.
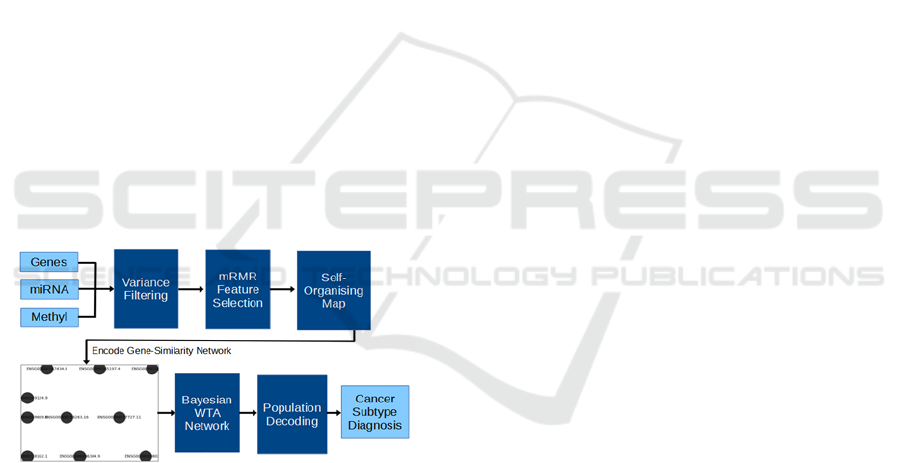
Figure 3: Diagram representing the methodological process
for this research. We start with multi-omic data, apply pre-
processing steps, and encode into a binary image. These are
used to train a Bayesian WTA network, the responses from
which we can then use various methods to decode into a
final prediction from the system.
4 EXPERIMENTAL RESULTS
In this section, we experimentally evaluate the
performance of a hierarchical Bayesian WTA network
on the TCGA datasets for the BRCA and KIRC cancer
subtypes. The network we choose for our
experimentation is based on the design presented in
Guo et al. (2017), making use of the code
implementation provided by Himst et al. (2023). The
network is composed of an input, hidden, and output
layer. The shape of the input layer is determined by
the pixel size of GSN images, which is 176 x 128. We
split the image into 16 subsections of size 11 x 8. Each
of these 16 sensory blocks then feeds into a layer of
WTA circuits with 32 hidden neurons. Finally, each
of the neurons in the hidden layer is connected
through a single WTA circuit consisting of 100 output
neurons. The network includes top-down connections
as suggested by Himst et al. (2023) in an effort to
improve the network’s learning and classification
performance. To evaluate the classification
performance of our methodologies, we use the metric
of F1 score. F1 score was chosen over the typical
accuracy metric for classification, as the TCGA
datasets contain heavy class imbalance. Due to the
nature of many population coding methods, it
becomes trivial to achieve a high accuracy score by
training a network which only predicts the majority
class regardless of the input. In fact, this is an
outcome which we must take steps to actively avoid
in some cases, such as by applying SMOTE to the
dataset. Furthermore, F1 score gives a higher
weighting to the classification performance of the
positive class, which is pertinent for cancer detection
due to the asymmetrical real-life ramifications of
reporting a False Negative versus False Positive
result. In the case of all experiments involving
SMOTE, the F1 score is reported only on data present
in the original dataset.
4.1 Effects of Imbalanced Datasets on
Population Coding
One of the key goals of this research is to highlight
how different methods of population coding respond
to class imbalance. In order to demonstrate this, we
use SMOTE to adjust the class ratio α of the training
sets for the WTA network, and perform K-folds cross
validation on the rebalanced datasets, retraining the
network each time. We can then test each of the
population decoding methodologies described in
Section 2. The results of this experiment are shown
in Figure 4. As we can see from Figure 4, the
performance of the population vector decoder has a
strong positive correlation with α. Not pictured in
Figure 4 is the winner-take-all decoder, which had a
consistent F1 score of zero both on each training fold
and over the entire dataset, regardless of α. In the case
of where no SMOTE was applied and the network is
trained on the standard TCGA dataset, both of these
methods report an F1 score of zero. Probing deeper,
BIOINFORMATICS 2024 - 15th International Conference on Bioinformatics Models, Methods and Algorithms
398
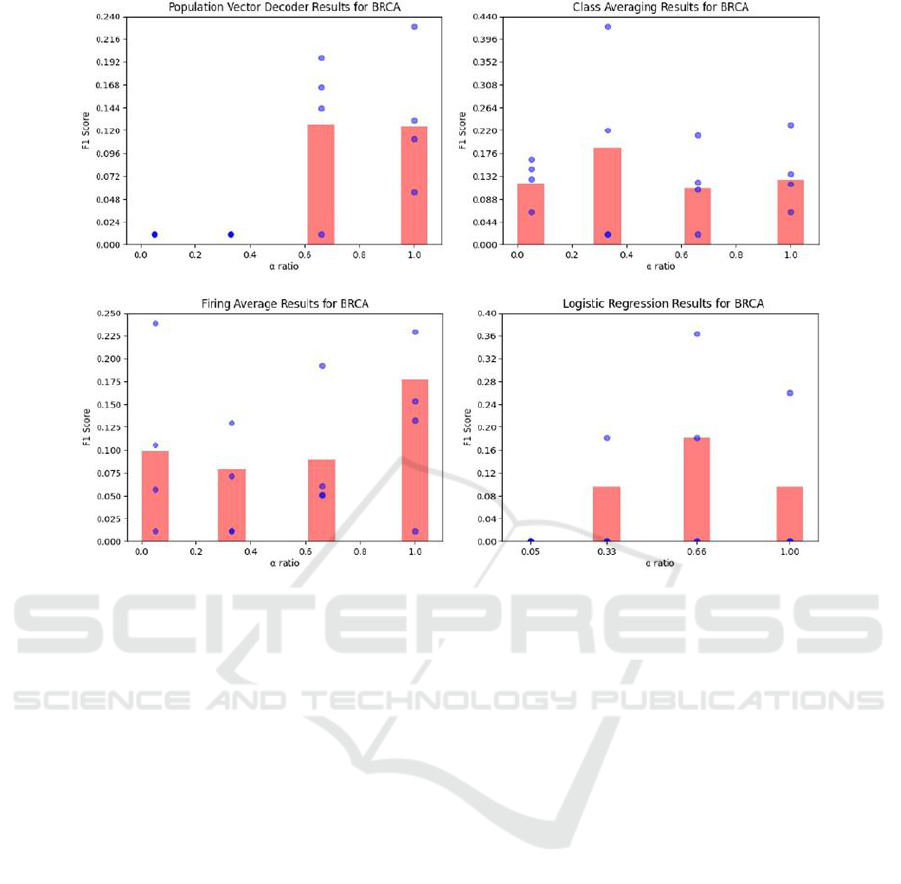
Figure 4: Results for different population decoding methods of SNNs trained on datasets with varying levels of synthetic
oversampling. Each red bar represents the score over the entire dataset, whereas each blue dot represents the score for a single
fold.
this is due to every neuron in the output population
being associated with the majority class, thereby
rendering the system unable to make predictions of
the minority class. Logistic regression had similar
issues with performance on the default dataset with a
low α ratio, but saw a marked improvement at the
point of oversampling to α=0.33 for BRCA and
α=0.66 for KIRC, and performed reasonably well
above these thresholds. Class averaging and firing
averaging both performed considerably better across
all α values. They therefore demonstrate resilience to
class imbalance, as there appears to be no strong
correlation between their predictive performance and
α ratio. Across both datasets, class averaging had
the highest single performance of any population
decoding method trialled in this research.
4.2 Distribution of Neuron Class
Assignments
Herein lies an exploration of neuron class
assignments. Presented in Figure 5 is a heatmap of the
α ratio of neuron class assignments determined via
Equation (2). We further calculate the Pearson
correlation coefficient between the α ratio of the
training dataset and the neuron class assignments:
The BRCA dataset has a dataset-neuron α correlation
coefficient of 0.932, and KIRC 0.879. Both datasets
showing such a strong correlation is certainly
indicative of the relationship between the distribution
of neuron classes and classes in the training set. This
result is notable as one may expect, for instance, that
the number of neurons assigned to a certain class be
dependent upon the complexity of the stimuli within
that class. Querlioz et al. (2013) ascribe the
improvement of predictive performance when
increasing the size of the population of output
neurons to the notion that the population is able to
learn more diverse representations of the output class.
However, as our chosen SMOTE technique is to
simply oversample the minority class, complexity of
the stimuli is constant across varying levels of α ratio.
From these results, we suggest that the determining
factor for the class assignment would therefore appear
to be the class distribution of the stimulus set. Another
point of interest is that whilst the correlation between
the α ratios is high, the heatmap in Figure 5 shows
that the relationship is not exactly linear. On the
unmodified datasets, the low α ratio leads to mode
collapse, where every neuron in the output population
Neural Population Decoding and Imbalanced Multi-Omic Datasets for Cancer Subtype Diagnosis
399
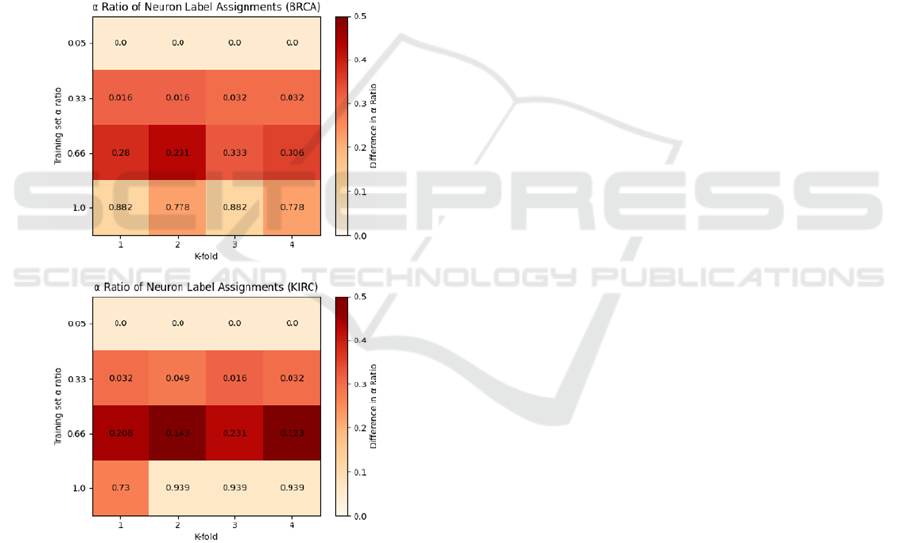
is assigned to the majority class. For the highest α
ratio, where the dataset was synthetically
oversampled to have an equal class distribution, the
neuron class distribution also reaches a similarly high
α ratio. However, for both the α=0.33 and α=0.66
datasets, there is a much larger discrepancy between
the dataset and neuron assignments. This is likely
indicative of the shortcomings of Equations (1) & (2)
when dealing with class imbalance which we
introduced in Section 2 – regardless of whether a
neuron is presented with 3 or 6 examples from the
minority class, if it’s shown 10 from the majority
class then it has a considerably greater likelihood of
being assigned as a majority neuron. This hypothesis
further explains the “jump” in neuron assignments
when moving from α=0.66 to α=1.0, as the minority
class is finally placed on equal footing with the
majority class.
Figure 5: Heatmap of the relationship between the α ratio
of neuron class assignments versus the class distribution of
the training set. The numerical value within each cell is the
α ratio of neuron assignments for each of the K-folds during
training. The colour scale represents the absolute difference
between the neuron assignment α ratio and the α ratio of
the training set.
4.3 Multi versus Single Omics
In this section, we analyse the network’s performance
when trained on subsets of the omic information
present in the BRCA dataset. The results of these
experiments are shown in Figure 6. Our results
generally concur with that of other researchers in
relation to the predictive power of the omic types
(Fatima & Rueda, 2020). Utilising all multi-omic
features together leads to the best classification
results. This is followed by genomic, mitochondrial
RNA, and methylation features (respectively). These
results are encouraging as they support the network’s
claim to be effectively learning information from the
input stimuli, despite the fact that the overall
classification is quite poor in comparison with other
techniques applied to TCGA datasets (Fatima &
Rueda, 2020).
4.4 Test Set Bias
Here, we demonstrate the effects of the bias
introduced by performing population decoding using
the responses to a test set, and then predicting on that
same test set. First, consider using a trained WTA
network to perform classification upon a test set,
using any of the population decoding methods
defined in Section 2 where Z is a prerequisite (i.e. not
logistic regression). In this example, we set the initial
size of the test set to only one stimulus sample.
Following Equation (1) to construct Z, we notice that
it is impossible for the system to produce an incorrect
prediction; whichever neuron(s) spiked when
presented with the stimulus are retroactively assigned
to be a neuron of that class. Shown in Figure 6 is the
accuracy score when using a completely untrained
network’s responses over various sized subsets of the
testing set to determine neuron class associations. We
use accuracy score in this case rather than F1, as the
randomly distributed test subsets may not contain any
positive samples, thereby making F1 inapplicable. We
can clearly see from Figure 7 that the “infallible
neuron” problem arises when the size of the test set
has a low number of samples. We can further
determine that the bias introduced by this
methodology decreases as the number of test samples
increases. This is likely why the issue was not
identified by Himst et al. (2023) & Guo et al. (2017)
when working with the 10,000 sample MNIST testing
set, but is certainly something to be wary of with
smaller datasets. We posit that this may also be a
contributing factor to the ability of these systems to
have such high performance whilst only being
exposed to the training set for one epoch, whereas
Nessler et al. (2013), Diehl & Cook (2015) and
Querlioz et al. (2013) all required multiple epochs to
achieve similar results. Luckily, the solution we
propose to address this bias is extremely simple: use
BIOINFORMATICS 2024 - 15th International Conference on Bioinformatics Models, Methods and Algorithms
400
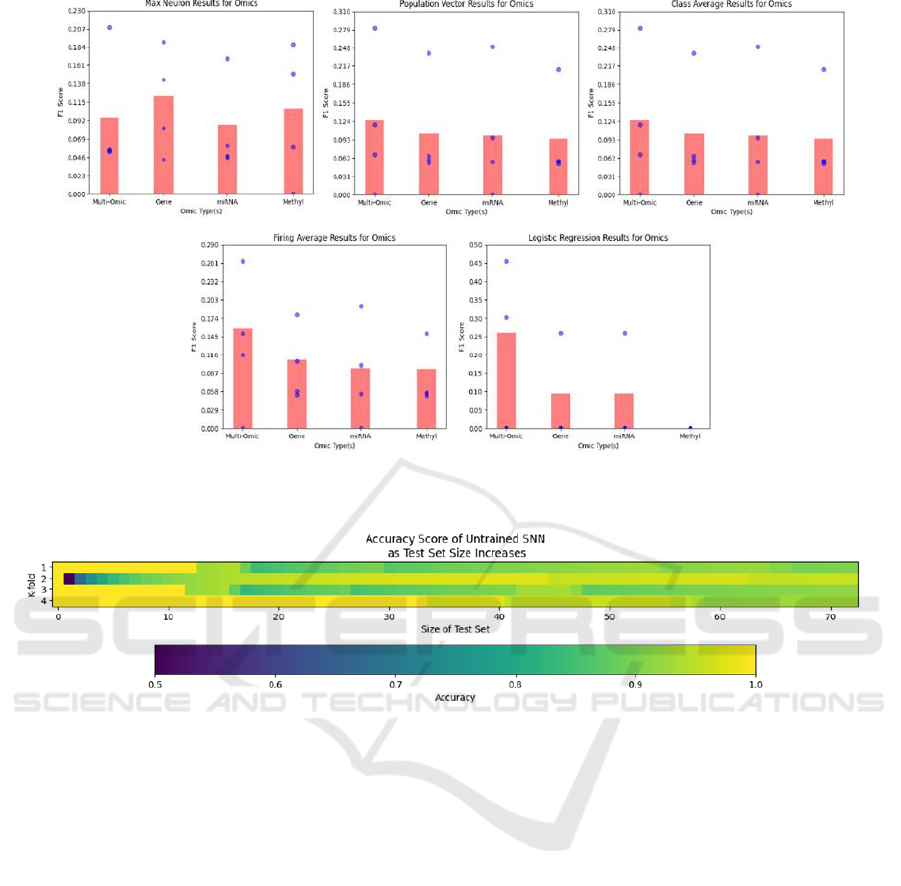
Figure 6: F1 score results for the WTA network trained on various subsets of omic information using K-folds cross validation.
Each bar represents the score over the entire dataset, whereas each blue dot represents the score for a single fold.
Figure 7: Heatmap of the accuracy score results of an untrained SNN making predictions upon a test set of increasing size,
where the population decoding is calculated using the responses of the network to that same test. The results shown here are
calculated over the BIRC dataset split into 4 folds.
the network responses from the training set to
perform population decoding, rather than the test set
responses. This removes the need to use test labels to
make predictions, and importantly eliminates the
infallible neuron problem. One additional piece of
advice for practitioners making this change is to only
start record training responses after the network has
been trained for a desired number of epochs, as using
the responses from an untrained network is liable to
give poor results. Diehl & Cook (2015) show that
following these practices can still lead to strong
classification performance.
5 CONCLUSION
Throughout this work, we have explored the
motivation and implications of implementing an array
of both common and novel population decoding
strategies for multi-omic based cancer subtype
diagnosis. Our findings show that the winner-take-all
and population vector decoders are both heavily
impacted by class imbalance, whereas class
averaging, logistic regression, and our novel firing
average implementation are more imbalance
resistant. We further show that the assignment of
neuron classes in population decoders is highly
correlated with the class distribution of the stimulus
set. This is a property which has, as far as we can
identify, gone unnoted thus far in the literature, and
warrants further investigation into the relationship
between the complexity of stimuli, class distribution
and neuron assignments.
Overall, the predictive performance of our system
is poor in comparison to other methods using this
dataset (Fatima & Rueda, 2020). In our estimation,
the primary issue lies in the loss of information when
transforming multi-omic data into binarized GSN
Neural Population Decoding and Imbalanced Multi-Omic Datasets for Cancer Subtype Diagnosis
401

images. We can observe from Figure 1 that a simple
logistic regression model is capable of achieving
near-perfect results using the same feature subset,
thereby isolating the problem to either the GSN or the
SNN. We later tested the classification performance
of a simple Convolutional Neural Network (CNN) on
the GSN images, which likewise had difficulty
extracting information from the binary data, and
surprisingly lead to even poorer predictive
performance than the SNN system. Thus, we
conclude that more sophisticated methods of
information encoding are necessary if we wish to
apply SNNs to datasets of within the domain of multi-
omics, or indeed to continuous tabular datasets in
general.
Despite the quality of our predictions, we are
nevertheless able to identify issues with current
practices in regards to the bias introduced by utilising
testing set labels and responses to perform population
decoding. Our recommendation for future researchers
is that this practice be avoided in favour of using the
training set, with the caveat that the network be
trained for at least one epoch before collecting the
responses.
Several challenges that we faced during this
research were related to the highly stochastic nature
of Bayesian WTA networks. This leads to high
variance in training convergence, making the
system’s performance difficult to evaluate in general
terms. K-folds cross validation is absolutely
necessary in this instance to gain insight into the
variance of results between runs. Furthermore, due to
their non-parallelizable nature and high
dimensionality requirements, training times can be
exceedingly long (Querlioz et al. (2013) report
approximately 8 hours for one run on the MNIST
dataset). Combined, these two factors make iterative
improvement difficult, as well as making it intractable
to explore high dimensional hyperparameter spaces.
One area for future research could therefore be the
application of more computing power to properly
perform hyperparameter optimisation on the network,
which could lead to superior performance.
Future research may alternatively wish to focus on
a biologically plausible method of translating
population responses into discrete decisions. One
potential direction for this is suggested by Hao et al.
(2020), wherein they combine the unsupervised
STDP learning rule with a leaky integrate-and-fire
neuron model to perform classification on the MNSIT
dataset. We have identified the encoding of
information as a bottleneck, as SNNs necessitate the
discretisation of information into spikes, inherently
impairing the maximum information density of the
system. To perhaps alleviate this problem, further
research could attempt alternative spike encoding
methods, such as those suggested by Guo et al.
(2021). On the other end of the system, interesting
insights could be gained by investigating the temporal
nature of neural responses, as opposed to the spike
count code (Grün & Rotter, 2010).
REFERENCES
Yamazaki K, Vo-Ho VK, Bulsara D, Le N. Spiking Neural
Networks and Their Applications: A Review. Brain Sci.
2022 Jun 30;12(7):863.
van der Himst, O., Bagheriye, L., & Kwisthout, J. (2023,
August). Bayesian Integration of Information Using
Top-Down Modulated WTA Networks. arXiv e-prints,
arXiv:2308.15390. doi: 10.48550/arXiv.2308.15390
Ma WJ, Pouget A. Population codes: Theoretic aspects.
Encyclopedia of neuroscience. 2009 Jun;7:749-55.
Guo, S., Yu, Z., Deng, F., Hu, X., & Chen, F. (2017).
Hierarchical bayesian inference and learning in
spiking neural networks. IEEE transactions on
cybernetics, 49 (1), 133–145.
Grün, S. and Rotter, S. eds., 2010. Analysis of parallel spike
trains (Vol. 7). Springer Science & Business Media.
J. M. Beck et al., Probabilistic population codes for
Bayesian decision making, Neuron, vol. 60, no. 6, pp.
1142–1152, 2008.
Shamir, M. (2009, 02). The temporal winner-take-all
readout. PLOS Computational Biology, 5 (2), 1-13.
doi:10.1371/journal.pcbi.1000286.
D. Kersten, P. Mamassian, and A. Yuille, Object perception
as bayesian inference, Annual Review of Psychology,
vol. 55, no. 1, pp. 271–304, 2004, pMID: 14744217.
T. S. Lee and D. Mumford, Hierarchical Bayesian inference
in the visual cortex, J. Opt. Soc. America A Opt. Image
Sci. Vis., vol. 20, pp. 1434–1448, 2003.
K. Friston, The free-energy principle: A unified brain
theory?, Nat. Rev. Neurosci., vol. 11. 127–138, 2010.
B. Nessler, M. Pfeiffer, L. Buesing, W. Maass, Bayesian
computation emerges in generic cortical microcircuits
through spiketiming-dependent plasticity, PLoS
Comput. Biol., vol. 9, no. 4, 2013, Art. no. e1003037.
P. U. Diehl and M. Cook, Unsupervised learning of digit
recognition using spike-timing-dependent plasticity,
Front. Comput. Neurosci., vol. 9, p. 99, Aug. 2015
Querlioz, Damien & Bichler, Olivier & Dollfus, Philippe &
Gamrat, Christian. (2013). Immunity to Device
Variations in a Spiking Neural Network With
Memristive Nanodevices. IEEE Transactions on
Nanotechnology. 12. 288-295.
10.1109/TNANO.2013.2250995.
Hao, Y., Huang, X., Dong, M., & Xu, B. (2020). A
biologically plausible supervised learning method for
spiking neural networks using the symmetric stdp rule.
Neural Networks, 121, 387-395 doi:
https://doi.org/10.1016/j.neunet.2019.09.007.
BIOINFORMATICS 2024 - 15th International Conference on Bioinformatics Models, Methods and Algorithms
402

Weinstein, J.N.Collisson, E. A. Mills, G. B.Shaw, K.R.
Ozenberger, B.A.Stuart (2013, October). The cancer
genome atlas pan-cancer analysis project, Cancer
Genome Atlas Research Network, Nat Genet, 45 (10),
1113-1120.
Fatima, N., & Rueda, L. (2020, 05). iSOM-GSN: an
integrative approach for transforming multi-omic data
into gene similarity networks via self-organizing maps.
Bioinformatics, 36 (15), 4248-4254.
Ang, JC & Mirzal, Andri & Haron, Habibollah & Hamed,
Haza Nuzly Abdull. (2015). Supervised, Unsupervised,
and Semi-Supervised Feature Selection: A Review on
Gene Selection. IEEE/ACM Transactions on
Computational Biology and Bioinformatics. 13.
Ding C, Peng H. Minimum redundancy feature selection
from microarray gene expression data. J Bioinform
Comput Biol. 2005;3(2):185-205.
T. Kohonen, The self-organizing map, in Proceedings of the
IEEE, vol. 78, no. 9, pp. 1464-1480, Sept. 1990, doi:
10.1109/5.58325.
Guo, Wenzhe & Fouda, Mohammed E. & Eltawil, Ahmed
& Salama, Khaled. (2021). Neural Coding in Spiking
Neural Networks: A Comparative Study for Robust
Neuromorphic Systems. Frontiers in Neuroscience. 15.
638474. 10.3389/fnins.2021.638474.
Chawla, Nitesh & Bowyer, Kevin & Hall, Lawrence &
Kegelmeyer, W. (2002). SMOTE: Synthetic Minority
Over-sampling Technique. J. Artif. Intell. Res. (JAIR).
16. 321-357. 10.1613/jair.953.
Imbalanced-learn: SMOTE. (2016). Imbalanced-learn.
Retrieved September 30, 2023.
Neural Population Decoding and Imbalanced Multi-Omic Datasets for Cancer Subtype Diagnosis
403
