
Integrating Structure and Sequence: Protein Graph Embeddings via
GNNs and LLMs
Francesco Ceccarelli
1
, Lorenzo Giusti
2
, Sean B. Holden
1
and Pietro Li
`
o
1
1
Department of Computer Science and Technology , University of Cambridge, Cambridge, U.K.
2
Department of Computer, Control and Management Engineering, Sapienza University, Rome, Italy
Keywords:
Graph Neural Networks, Large Language Models, Protein Representation Learning.
Abstract:
Proteins perform much of the work in living organisms, and consequently the development of efficient com-
putational methods for protein representation is essential for advancing large-scale biological research. Most
current approaches struggle to efficiently integrate the wealth of information contained in the protein sequence
and structure. In this paper, we propose a novel framework for embedding protein graphs in geometric vector
spaces, by learning an encoder function that preserves the structural distance between protein graphs. Utiliz-
ing Graph Neural Networks (GNNs) and Large Language Models (LLMs), the proposed framework generates
structure- and sequence-aware protein representations. We demonstrate that our embeddings are successful
in the task of comparing protein structures, while providing a significant speed-up compared to traditional
approaches based on structural alignment. Our framework achieves remarkable results in the task of protein
structure classification; in particular, when compared to other work, the proposed method shows an average
F1-Score improvement of 26% on out-of-distribution (OOD) samples and of 32% when tested on samples
coming from the same distribution as the training data. Our approach finds applications in areas such as drug
prioritization, drug re-purposing, disease sub-type analysis and elsewhere.
1 INTRODUCTION
Proteins are organic macro-molecules made up of
twenty types of natural amino acids. Almost all in-
teractions and reactions which occur in living organ-
isms, from signal transduction, gene transcription and
immune function to catalysis of chemical reactions,
involve proteins (Morris et al., 2022). The compari-
son of proteins and their structures is an essential task
in bioinformatics, providing support for protein struc-
ture prediction (Kryshtafovych et al., 2019), the study
of protein-protein docking (Lensink et al., 2018),
structure-based protein function prediction (Gherar-
dini and Helmer-Citterich, 2008) and many further
tasks. Considering the large quantity of protein data
stored in the Protein Data Bank (PDB) (Berman et al.,
2003) and the rapid development of methods for per-
forming protein structure prediction (for example, Al-
phaFold2 (Jumper et al., 2021)), it is desirable to de-
velop methods capable of efficiently comparing the
tertiary structures of proteins.
Generally, protein comparison methods can be
divided into two classes: alignment-based meth-
ods (Akdel et al., 2020; Shindyalov and Bourne,
1998; Kihara and Skolnick, 2003) and alignment-free
methods (Xia et al., 2022; Røgen and Fain, 2003;
Budowski-Tal et al., 2010; Zotenko et al., 2006). The
former aim at finding the optimal structural super-
position of two proteins. A scoring function is then
used to measure the distance between each pair of
superimposed residues. For such methods (for ex-
ample (Holm and Sander, 1993; Zhang and Skol-
nick, 2005)) the superposition of the atomic structures
is the main bottleneck as it has been proven to be
an NP-hard problem (Lathrop, 1994). On the other
hand, alignment-free methods try to represent each
protein in the form of a descriptor, and then to mea-
sure the distance between pairs of descriptors (Xia
et al., 2022). Descriptors need to satisfy two require-
ments: (1) their size should be fixed and independent
of the length of proteins; (2) they should be invariant
to rotation and translation of proteins.
The template modeling score (TM-score) (Zhang
and Skolnick, 2004) is a widely used metric for as-
sessing the structural similarity between two pro-
teins. It is based on the root-mean-square deviation
(RMSD) of the atomic positions in the proteins, but
considers the lengths of the proteins and the number
582
Ceccarelli, F., Giusti, L., Holden, S. and Liò, P.
Integrating Structure and Sequence: Protein Graph Embeddings via GNNs and LLMs.
DOI: 10.5220/0012453600003654
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 13th International Conference on Pattern Recognition Applications and Methods (ICPRAM 2024), pages 582-593
ISBN: 978-989-758-684-2; ISSN: 2184-4313
Proceedings Copyright © 2024 by SCITEPRESS – Science and Technology Publications, Lda.

of residues that can be superimposed. TM-score has
been shown to be highly correlated with the similar-
ity of protein structures and can be used to identify
structurally similar proteins, even when they have low
sequence similarity. Unfortunately, computing TM-
scores is computationally intractable even for rela-
tively small numbers of proteins. TM-align (Zhang
and Skolnick, 2005), one of the popular alignment-
based methods, takes about 0.5 seconds for one struc-
tural alignment on a 1.26 GHz PIII processor. As
such, computing TM-scores for existing databases,
containing data for millions of proteins, is unafford-
able. While several deep learning methods for protein
comparison have been developed (for example, Deep-
Fold (Liu et al., 2018) and GraSR (Xia et al., 2022))
they suffer from major drawbacks: (1) they are trained
by framing the protein comparison task as a classi-
fication problem—that is, predicting if two proteins
are structurally similar—and hence fail to directly in-
corporate TM-scores in the loss function formulation;
(2) they produce latent representations (embeddings)
which do not integrate the information contained in
the protein sequences and structures; (3) they usually
do not exploit the inductive bias induced by the topol-
ogy of graph-structured proteins, and they fail to con-
sider different geometries of the latent space to match
well the underlying data distribution.
In this paper, we address the aforementioned lim-
itations of current protein embedding methods by
proposing an efficient and accurate technique that in-
tegrates both protein sequence and structure infor-
mation. In detail, we first construct protein graphs
where each node represents an amino acid in the pro-
tein sequence. We then generate features for each
amino acid (node in the graph) using Large Language
Models (LLMs) before applying Graph Neural Net-
works (GNNs) to embed the protein graphs in geo-
metric vector spaces while combining structural and
sequence information. By incorporating TM-scores
in the formulation of the loss function, the trained
graph models are able to learn a mapping that pre-
serves the distance between the input protein graphs,
providing a way to quickly compute similarities for
every pair of unseen proteins. We evaluated the pro-
posed approach and its ability to generate meaning-
ful embeddings for downstream tasks on two protein
datasets. On both, the proposed approach reached
good results, outperforming other current state-of-
the-art methods on the task of structural classification
of proteins on the SCOPe dataset (Fox et al., 2014).
Contribution. The main contributions of this paper
can be summarised as follow: (i) A novel learning
framework for generating protein representations in
geometric vector spaces by merging structural and se-
quence information using GNNs and LLMs. (ii) A
quick and efficient method for similarity computation
between any pair of proteins. (iii) An evaluation of
the ability of our embeddings, in both supervised and
unsupervised settings, to solve downstream protein
classification tasks, and a demonstration of their su-
perior performance when compared to current state-
of-the-art methods. Our approach finds a plethora of
applications in the fields of bioinformatics and drug
discovery.
2 BACKGROUND AND RELATED
WORK
Several alignment-based methods have been proposed
over the years, each exploiting different heuristics to
speed up the alignment process. For example, in
DALI (Holm and Sander, 1999), Monte Carlo opti-
mization is used to search for the best structural align-
ment. In Shindyalov and Bourne (1998), the authors
proposed combinatorial extension (CE) for similarity
evaluation and path extension. An iterative heuristic
based on the Needleman–Wunsch dynamic program-
ming algorithm (Needleman and Wunsch, 1970) is
employed in TM-align (Zhang and Skolnick, 2005),
SAL (Krishna et al., 1997) and STRUCTAL (Zhang
and DeLisi, 1997). Examples of alignment-free ap-
proaches are Scaled Gauss Metric (SGM) (Røgen
and Fain, 2003) and the Secondary Structure Element
Footprint (SSEF) (Zotenko et al., 2006). SGM treats
the protein backbone as a space curve to construct a
geometric measure of the conformation of a protein,
and then uses this measure to provide a distance be-
tween protein shapes. SSEF splits the protein into
short consecutive fragments and then uses these frag-
ments to produce a vector representation of the pro-
tein structure as a whole. More recently, methods
based on deep learning have been developed for the
task of protein structure comparison. For instance,
DeepFold (Liu et al., 2018) used a deep convolu-
tional neural network model trained with the max-
margin ranking loss function (Wang et al., 2016) to
extract structural motif features of a protein, and learn
a fingerprint representation for each protein. Cosine
similarity was then used to measure the similiarity
scores between proteins. DeepFold has a large num-
ber of parameters, and fails to exploit the sequence in-
formation and the topology of graph-structured data.
GraSR (Xia et al., 2022) employs a contrastive learn-
ing framework, GNNs and a raw node feature extrac-
tion method to perform protein comparison. Com-
pared to GraSR, we present a general framework to
produce representations of protein graphs where the
Integrating Structure and Sequence: Protein Graph Embeddings via GNNs and LLMs
583

distance in the embedding space is correlated with the
structural distance measured by TM-scores between
graphs. Finally, our approach extends the work pre-
sented in Corso et al. (2021), which was limited to bi-
ological sequence embeddings, to the realm of graph-
structured data.
3 MATERIAL AND METHODS
The core approach, shown in Figure 1, is to map
graphs into a continuous space so that the distance be-
tween embedded points reflects the distance between
the original graphs measured by the TM-scores. The
main components of the proposed framework are the
geometry of the latent space, a graph encoder model,
a sequence encoder model, and a loss function. De-
tails for each are as follows.
3.1 Latent Space Geometry
The distance function used (d in Figure 1) defines the
geometry of the latent space into which embeddings
are projected. In this work we provide a comparison
between Euclidean, Manhattan, Cosine and squared
Euclidean (referred to as Square) distances (details in
Appendix B).
3.2 Graph Encoder Model
The encoder performs the task of mapping the in-
put graphs to the embedding space. A variety of
models exist for this task, including linear, Multi-
layer Perceptron (MLP), LSTM (Cho et al., 2014),
CNN (Fukushima, 1980) and Transformers (Vaswani
et al., 2017). Given the natural representation of pro-
teins as graphs, we chose GNNs as encoder mod-
els. We have constructed the molecular graphs of
proteins starting from PDB files. A PDB file con-
tains structural information such as 3D atomic coor-
dinates. Let G = (V, E) be a graph representing a pro-
tein, where each node v ∈ V is a residue and inter-
action between the residues is described by an edge
e ∈ E. Two residues are connected if they have any
pair of atoms (one from each residue) separated by
a Euclidean distance less than a threshold distance.
The typical cut-off, which we adopt in this work, is 6
angstroms (
˚
A) (Chen et al., 2021).
3.3 Sequence Encoder Model
Given the graph representation of a protein, each
node v of the graph (each residue) must be associ-
ated with a feature vector. Typically, features ex-
Table 1: Investigated node attributes and their dimensions.
BERT and LSTM features are extracted using LLMs pre-
trained on protein sequences (ProBert (Brandes et al., 2022)
and SeqVec (Heinzinger et al., 2019)).
Feature Dimension
One hot encoding of amino acids 20
Physicochemical properties 7
BLOcks SUbstitution Matrix 25
BERT-based language model 1024
LSTM-based language model 1024
tracted from protein sequences by means of LLMs
have exhibited superior performances compared to
handcrafted features. We experimented with five dif-
ferent sequence encoding methods: (1) a simple one-
hot encoding of each residue in the graph, (2) seven
physicochemical properties of residues as extracted
by Meiler et al. (2001), which are assumed to in-
fluence the interactions between proteins by creat-
ing hydrophobic forces or hydrogen bonds between
them, (3) the BLOcks SUbstitution Matrix (BLO-
SUM) (Henikoff and Henikoff, 1992), which counts
the relative frequencies of amino acids and their sub-
stitution probabilities, (4) features extracted from pro-
tein sequences employing a pre-trained BERT-based
transformer model (ProBert (Brandes et al., 2022)),
and (5) node features extracted using a pre-trained
LSTM-based language model (SeqVec (Heinzinger
et al., 2019)). Table 1 summarizes the node features
and their dimensions, while Figure 2 depicts the pro-
cess of constructing a protein graph with node fea-
tures, starting from the corresponding protein data.
3.4 Loss Function
The loss function used, which minimises the MSE be-
tween the graph distance and its approximation as the
distance between the embeddings, is
L =
∑
g
1
,g
2
∈G
(TM(g
1
, g
2
) −d (GNN
θ
(g
1
), GNN
θ
(g
2
)))
2
(1)
where G is the training set of protein graphs,
GNN
θ
is the graph encoder and θ represents the pa-
rameters of the model. The TM-score is a similarity
metric in the range (0,1], where 1 indicates a perfect
match between two structures. Since the formulation
of the loss is expressed in terms of distances, we re-
formulate the TM-scores as a distance metric by sim-
ply computing TM(g
1
, g
2
) = 1 − TM
score
(g
1
, g
2
). By
training neural networks to minimize the loss in Equa-
tion 1, we encourage the networks to produce latent
representations such that the distance between these
representations is proportional to the structural dis-
tance between the input graphs.
ICPRAM 2024 - 13th International Conference on Pattern Recognition Applications and Methods
584

d
Structural !
Distance
Input proteins Protein Graphs with sequence attributes
Graph Encoder
3-Dimensional Latent Space
of Protein Graphs
Structural comparison
Protein structural
classification
Protein function
prediction
Example applications of protein
embeddings
Figure 1: We learn an encoder function that preserves the structural distance, measured by the TM-score, between two input
proteins. We construct protein graphs by combining sequence and structure information as shown in Figure 2. A distance
function d defines the shape of the latent space. The generated embeddings can be used for a variety of applications in
bioinformatics and drug discovery. (For simplicity, this Figure depicts a 3-dimensional latent space.).
Figure 2: Graph representation of a protein, which com-
bines sequence and structure. Starting from protein data (a
PDB file from, for example, UniProt or PDB), we extract
protein sequence and structure information. We construct
graphs where each node represents an amino acid in the pro-
tein sequence. We then generate features for each node in
the graph using Large Language Models pre-trained on pro-
tein sequences.
4 PROTEIN DATASETS
We evaluated the proposed approach on two pro-
tein datasets. First, we downloaded the human pro-
teome from UniProt
1
and sub-selected 512 protein
kinases. To obtain the TM-scores to train the graph
models, we evaluated the structural similarity using
TM-align (Zhang and Skolnick, 2005). All-against-
all alignment yielded a dataset composed of 130,816
total comparisons. Every kinase in the dataset is cat-
egorized in one of seven family groups: (a) AGC
(63 proteins), (b) CAMK (82 proteins), (c) CK1 (12
proteins), (d) CMGC (63 proteins), (e) STE (48 pro-
teins), (f) TK (94 proteins), and (g) TKL (43 pro-
teins). The number of nodes in the graphs ranges from
1
https://www.uniprot.org
253 to 2644, with an average size of approximately
780 nodes. The average degree in the graphs is ap-
proximately 204, the average diameter of the graphs
is approximately 53 nodes and the maximum diam-
eter is 227 nodes. We further used the 40% iden-
tity filtered subset of SCOPe v2.07 (March 2018) as
a benchmark dataset (Fox et al., 2014). This dataset
contains 13,265 protein domains classified in one of
seven classes: (a) all alpha proteins (2286 domains),
(b) all beta proteins (2757 domains), (c) alpha and
beta proteins (a/b) (4148 domains), (d) alpha and beta
proteins (a+b) (3378 domains), (e) multi-domain pro-
teins (alpha and beta) (279 domains), (f) membrane
and cell surface proteins and peptides (213 domains),
and g) small proteins (204 domains). We again used
TM-align with all-against-all settings to construct a
dataset of approximately 170 millions comparisons.
To reduce the computational time and cost during
training, we randomly sub-sampled 100 comparisons
for each protein to create a final dataset of 1,326,500
comparisons. For this dataset, the number of nodes
in the graphs ranges from 30 to 9800, with an aver-
age size of approximately 1978 nodes. The average
degree is approximately 90, the average diameter of
the graphs is approximately 9 nodes and the maxi-
mum diameter is 53 nodes. Compared to benchmark
graph datasets (for example Sterling and Irwin (2015)
and Dwivedi et al. (2022)) we evaluated our approach
on graphs of significantly larger size (84 and 13 times
more nodes than the molecular graphs in Sterling and
Irwin (2015) and in Dwivedi et al. (2022), respec-
tively).
Integrating Structure and Sequence: Protein Graph Embeddings via GNNs and LLMs
585
5 EXPERIMENTAL RESULTS
5.1 Experimental Settings
We evaluate the proposed framework using Graph
Convolutional Networks (GCNs) (Kipf and Welling,
2016), Graph Attention Networks (GATs) (Veli
ˇ
ckovi
´
c
et al., 2017), and GraphSAGE (Hamilton et al., 2017)
(Appendix A). All the models were implemented
with two graph layers in PyTorch geometric (Fey and
Lenssen, 2019) to learn protein embeddings of size
256. Adam optimizer (Kingma and Ba, 2014) with
a learning rate of 0.001 was used to train the mod-
els for 100 epochs with a patience of 10 epochs. The
batch size was set to 100. We used 4 attention heads
in the GAT architecture. For each model, Rectified
Linear Units (ReLUs) (Nair and Hinton, 2010) and
Dropout (Srivastava et al., 2014) were applied after
each layer, and mean pooling was employed as read-
out function to obtain graph-level embeddings from
the learned node-level representations. Finally, each
experiment was run with 3 different seeds to provide
uncertainty estimates.
5.2 Kinase Embeddings
For the generation of the embeddings, we used 80%
of the kinase proteins for training and the remaining
20% for testing. Table 2
shows the MSE values for the graph encoders, us-
ing different choices of distance functions and node
features. For each model, the best scores are con-
sistently reached with LSTM-extracted features and
Euclidean geometry of the embedding space. Across
all models, GAT embeddings exhibit the lowest MSE,
followed by GarphSAGE and GCN. From Table 2, it
is clear that using pre-trained language models to ex-
tract node features from protein sequences leads to
better results. MSE scores for all distances across
all encoder models are lower when using BERT and
LSTM features. Furthermore, the LSTM-extracted
features perform consistently better compared to the
BERT ones. BLOSUM and Physicochemical features
are also usually associated with higher MSE for all
distances and models, indicating that they are poorly
correlated to TM-scores.
5.3 Fast Inference of TM-Scores
We employed the trained GAT architectures from Ta-
ble 2 to predict the TM-scores for the kinase pairs in
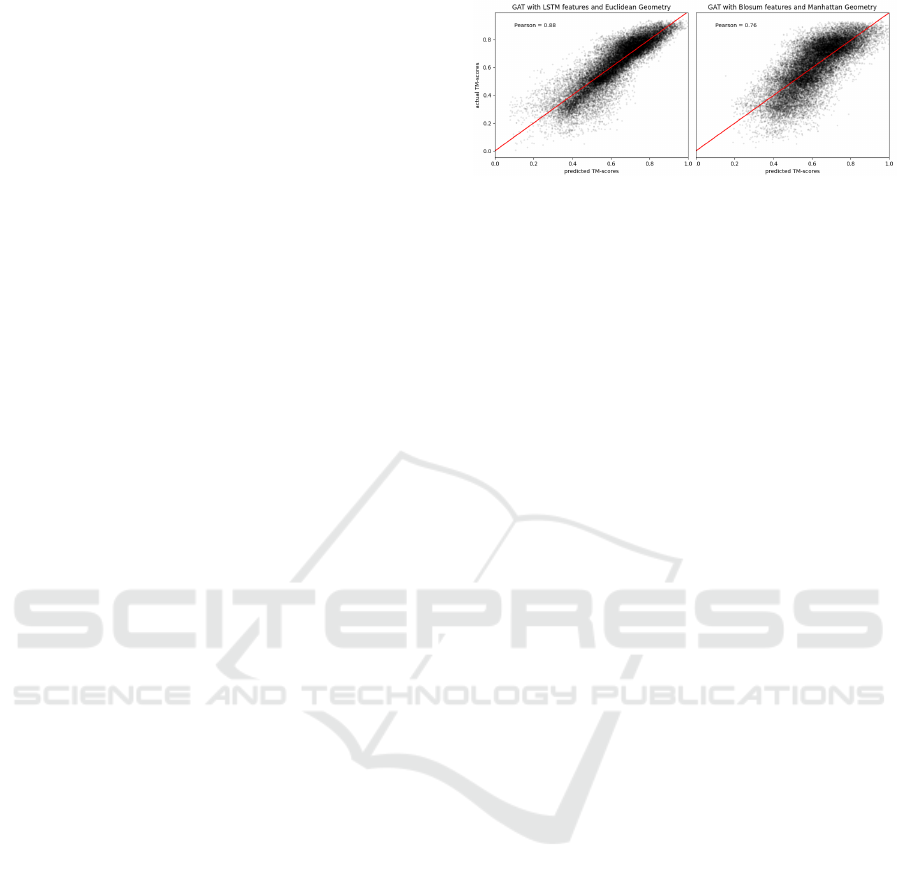
the test set. In Figure 3, we show the predicted versus
actual TM-scores for two combinations of features
and embedding geometries. The left plot in Figure 3
Figure 3: Actual versus predicted TM-scores. Using LSTM
features and Euclidean geometry (left) results in predictions
which follow more tightly the red line of the oracle com-
pared to BLOSUM features in the Manhattan space (right).
uses LSTM-extracted features and Euclidean space,
while the right one shows predictions for BLOSUM
features and Manhattan space. The complete quan-
titative evaluations, measured by Pearson correlation
between model predictions and true TM-scores for all
distances and features, are reported in Appendix D.
As in Table 2, the best performances are reached when
employing LSTM and BERT features while BLO-
SUM and Physicochemical features lead to the poor-
est performances (Appendix D). The highest correla-
tion score, reflecting the results reported in Table 2,
is reached when employing LSTM features and Eu-
clidean distance (Figure 3). It is worth noticing that,
for the 26,164 comparisons in the test set, the pro-
posed approach took roughly 120 seconds to compute
TM-scores. Executing TM-align with the same num-
ber of comparisons took 57,659 seconds (≈ 16 hours).
Details of the TM-score inference times for all the
models are given in Appendix D. The major speed-
up provided by performing inference using machine
learning models makes the proposed approach appli-
cable to datasets comprising millions of proteins.
5.4 Ablation Study: Structure Removal
Coupling GNNs with LLMs provides a means of in-
tegrating the information coming from the structure
and sequence of proteins. To analyse the benefits of
exploiting the topology induced by the graph struc-
tures, we performed an ablation study which disre-
gards such information. DeepSet (Zaheer et al., 2017)
considers objective functions defined on sets, that are
invariant to permutations. Using a DeepSet formu-
lation, we constructed protein graphs with features
where each node is only connected to itself. As for
the graph models, we trained DeepSet to minimize
the loss function in Equation 1 and report the results
in Table 3. Similarly to Table 2, the best MSE scores
are reached when using LSTM features and Euclidean
geometry. The scores in Table 3, computed by disre-
garding the graph connectivity and neighborhood in-
formation, are significantly higher than those reported
ICPRAM 2024 - 13th International Conference on Pattern Recognition Applications and Methods
586
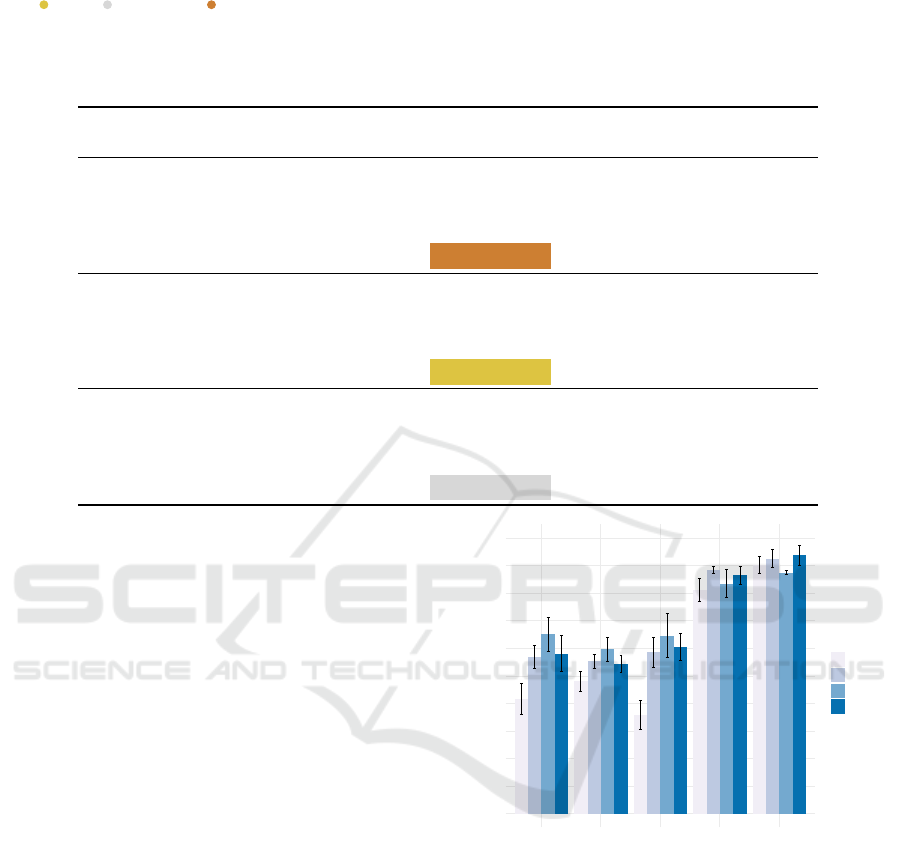
Table 2: MSE results for different feature types, distance functions and graph encoder models on the kinase dataset. We use
gold , silver , and bronze colors to indicate the first, second and third best performances, respectively. For each model,
the best scores are consistently reached with LSTM-extracted features and Euclidean geometry of the embedding space.
Across all models, GAT embeddings exhibit the best performance. For all the models, MSE scores are lower for features
extracted by means of LLMs (BERT and LSTM) compared to handcrafted feature extraction methods (one-hot, biochemical
and BLOSUM).
Model Feature Distance
Cosine Euclidean Manhattan Square
GCN
One hot 0.0194 ± 0.002 0.0380 ± 0.003 0.0192 ± 0.001 0.0729 ± 0.004
Physicochemical 0.0343 ± 0.012 0.0483 ± 0.009 0.0397 ±0.003 0.1109 ±0.007
BLOSUM 0.0327 ± 0.071 0.0271 ±0.043 0.0450±0.013 0.0697±0.023
BERT 0.0110 ± 0.003 0.0103 ± 0.001 0.0131 ± 0.006 0.0138 ±0.009
LSTM 0.0105 ±0.002 0.0088±0.004 0.0156 ± 0.001 0.0107 ± 0.004
GAT
One hot 0.0171 ± 0.001 0.0320 ± 0.012 0.0171 ± 0.011 0.0758 ± 0.009
Physicochemical 0.0295 ± 0.007 0.0328 ± 0.006 0.0220 ± 0.004 0.0856 ± 0.023
BLOSUM 0.0245 ± 0.012 0.0163 ± 0.009 0.0124 ± 0.011 0.0307 ± 0.009
BERT 0.0091 ± 0.018 0.0095 ± 0.008 0.0078 ± 0.009 0.0133 ± 0.011
LSTM 0.0088 ± 0.009 0.0073 ± 0.004 0.0086 ± 0.006 0.0101 ± 0.009
Graph
SAGE
One hot 0.0243 ± 0.002 0.0227 ± 0.011 0.0156 ± 0.009 0.0424 ± 0.010
Physicochemical 0.0301 ± 0.004 0.0266 ± 0.008 0.0310 ± 0.011 0.0578 ± 0.009
BLOSUM 0.0285 ± 0.007 0.0172 ± 0.008 0.0342 ± 0.002 0.0368 ± 0.007
BERT 0.0097 ± 0.011 0.0089 ± 0.007 0.0101 ± 0.007 0.0107 ± 0.009
LSTM 0.0093 ± 0.003 0.0084 ± 0.005 0.0143 ± 0.007 0.0094 ± 0.008
in Table 2 (p-value of t-test < 0.05 compared to GCN,
GAT and GraphSAGE). By considering patterns of
local connectivity and structural topology, GNNs are
able to learn better protein graph representations com-
pared to models which only exploit sequence-derived
features.
5.5 Downstream Task of Kinase
Classification
To prove the usefulness of the learned embeddings for
downstream tasks, we set out to classify each kinase
into one of the seven family groups (AGC, CAMK,
CK1, CMGC, STE, TK, TKL). Using the embeddings
generated by the GAT models, we trained an MLP,
composed of 3 layers of size 128, 64 and 32 respec-
tively, and a SoftMax classification head. The accu-
racy of classification, computed as the average result
of 5-fold cross-validation, for each feature type and
distance function is reported in Figure 4. The results
are consistent with Table 2: the best accuracies are
obtained when using LSTM- and BERT-extracted se-
quence features, while handcrafted feature extraction
methods (one hot, BLOSUM and physicochemical)
provide the poorest performance. The highest accu-
racy values of 93.7% and 92.48% are reached with
LSTM features and Square and Euclidean distance
functions, respectively.
0.0
0.2
0.4
0.6
0.8
1.0
One Hot Physicochemical Blosum BERT LSTM
Feature
Accuracy
Distance
Cosine
Euclidean
Manhattan
Square
Figure 4: Accuracy of classification for kinase family pre-
diction using the embeddings generated by the GAT models.
The highest accuracy value of 93.7% is reached with LSTM
features and the Square distance function.
5.6 Embedding out of Distribution
Samples
Being able to use pre-trained models for related or
similar tasks is essential in machine learning. We
tested the ability of the proposed graph models to
generalize to new tasks by generating embeddings for
the 13,265 proteins in the SCOPe dataset after being
trained only on kinase proteins. Given the better per-
formance provided by the use of LSTM features, in
this section we constructed protein graphs with LSTM
Integrating Structure and Sequence: Protein Graph Embeddings via GNNs and LLMs
587

Table 3: MSE values for an ablation study which disregards the topological information induced by the structure of the protein
graphs. We use gold , silver , and bronze colors to indicate the first, second and third best performances, respectively.
By ignoring the neighborhood and the structural information, the MSEs are significantly higher (p-value of t-test < 0.05)
compared to GNNs.
Model Feature Distance
Cosine Euclidean Manhattan Square
DeepSet
One Hot 0.1742 ± 0.003 0.0421 ± 0.002 0.0358 ± 0.001 0.0714 ± 0.003
Physicochemical 0.1766 ± 0.010 0.0437 ± 0.006 0.0464 ± 0.004 0.0900 ± 0.006
BLOSUM 0.1553 ± 0.003 0.0381 ± 0.009 0.0558 ± 0.008 0.0914 ± 0.008
BERT features 0.0132 ± 0.004 0.0129 ± 0.005 0.0192 ± 0.005 0.0220 ± 0.004
LSTM features 0.0141 ± 0.003 0.0116 ± 0.010 0.0348 ± 0.006 0.0200 ± 0.007
Table 4: Out of distribution (OOD) classification results on SCOPe proteins (F1-Score (OOD)). We use gold , silver , and
bronze colors to indicate the first, second and third best performances, respectively. Despite the different training data, the
GAT model with Euclidean and Square geometry outperforms all other approaches trained on SCOPe proteins. Classification
results for embeddings generated after training on SCOPe proteins are also shown (F1-Score); in this case, the proposed
approach outperforms the others by a larger margin for all choices of latent geometries.
Model Distance F1-Score (OOD) F1-Score
GAT
Cosine 0.6906 ± 0.0044 0.8290 ± 0.008
Euclidean 0.8204 ± 0.006 0.8557 ± 0.002
Manhattan 0.7055 ± 0.006 0.8481 ± 0.007
Square 0.8185 ± 0.004 0.8406 ± 0.006
SGM (Røgen and Fain, 2003) - - 0.6289
SSEF (Zotenko et al., 2006) - - 0.4920
DeepFold (Liu et al., 2018) - - 0.7615
GraSR (Xia et al., 2022) - - 0.8124
attributes and used a 3-Layer MLP as before to as-
sign the GAT-generated protein embeddings from the
SCOPe dataset to the correct class. Results of this
evaluation, measured as average F1-score across 5
folds for each distance function, are shown in Table 4
(F1-Score out of distribution (OOD)).
Euclidean and Square geometry of the embed-
ding space exhibited the best classification perfor-
mances. Despite being trained on OOD samples, the
proposed framework with Euclidean and Square ge-
ometry still managed to outperform the current state-
of-the-art results reported from models trained and
tested on SCOPe proteins, as shown in Table 4. The
superior performance, despite the different training
data, suggests the ability of the proposed approach to
learn meaningful protein representations by (1) merg-
ing structural and sequence information into a sin-
gle pipeline, and (2) capturing different and relevant
properties of the geometries of the latent space into
which embeddings are projected.
5.7 Protein Structural Classification
We constructed protein graphs with LSTM features
and trained the proposed GAT architectures on the
SCOPe dataset. The resulting MSE scores are re-
ported in Appendix D. The lowest score was again
reached when using Euclidean geometry for the latent
space. Using this model, we projected the protein em-
beddings onto two dimensions using t-SNE (Van der
Maaten and Hinton, 2008) as shown in Figure 5.
The high-level structural classes as defined in SCOPe
were captured by the proposed embeddings. While
not directly trained for this task, combining struc-
tural and sequence information allowed us to identify
small, local clusters representing the different protein
families in the SCOPe dataset. We employed super-
vised learning and trained a 3-layer MLP classifier
to label each protein embedding in the correct fam-
ily. Results of this evaluation, measured as average
F1-score across 5 folds, are shown in Table 4 (F1-
Score). When directly trained on SCOPe proteins, the
proposed approach outperforms the others by a large
margin for all choices of geometries (Table 4).
ICPRAM 2024 - 13th International Conference on Pattern Recognition Applications and Methods
588

Figure 5: t-SNE visualization of the learned embeddings,
coloured by protein structural family. The proposed ap-
proach generates protein embeddings which recapitulate the
different families in the SCOPe dataset.
6 CONCLUSION
In this paper, we presented a novel framework for
generating both structure- and sequence-aware pro-
tein representations. We mapped protein graphs with
sequence attributes into geometric vector spaces, and
showed the importance of considering different ge-
ometries of the latent space to match the underly-
ing data distributions. We showed that the gener-
ated embeddings are successful in the task of pro-
tein structure comparison, while providing an accu-
rate and efficient way to compute similarity scores
for large-scale datasets, compared to traditional ap-
proaches (Appendix D). The protein graph represen-
tations generated by our approach showed state-of-
the-art results for the task of protein structural clas-
sification on the SCOPe dataset. This work opens op-
portunities for future research, with potential for sig-
nificant contributions to the fields of bioinformatics,
structural protein representation and drug discovery
(Appendix E).
ACKNOWLEDGMENTS
For the purpose of open access, the author has applied
a Creative Commons Attribution (CC BY) licence to
any Author Accepted Manuscript version arising from
this submission. The protein structures in Figure 1
were downloaded from UniProt
2 3
under the Creative
Commons Attribution 4.0 International (CC BY 4.0)
License
4
and used without modifications.
2
https://www.uniprot.org
3
https://www.uniprot.org/help/license
4
https://creativecommons.org/licenses/by/4.0/
REFERENCES
Akdel, M., Durairaj, J., de Ridder, D., and van Dijk, A. D.
(2020). Caretta–a multiple protein structure alignment
and feature extraction suite. Computational and struc-
tural biotechnology journal, 18:981–992.
Berman, H., Henrick, K., and Nakamura, H. (2003). An-
nouncing the worldwide protein data bank. Nature
Structural & Molecular Biology, 10(12):980–980.
Brandes, N., Ofer, D., Peleg, Y., Rappoport, N., and Linial,
M. (2022). ProteinBERT: a universal deep-learning
model of protein sequence and function. Bioinformat-
ics, 38(8):2102–2110.
Budowski-Tal, I., Nov, Y., and Kolodny, R. (2010). Frag-
Bag, an accurate representation of protein structure,
retrieves structural neighbors from the entire PDB
quickly and accurately. Proceedings of the National
Academy of Sciences, 107(8):3481–3486.
Chen, J., Zheng, S., Zhao, H., and Yang, Y. (2021).
Structure-aware protein solubility prediction from se-
quence through graph convolutional network and pre-
dicted contact map. Journal of cheminformatics,
13(1):1–10.
Cho, K., Van Merri
¨
enboer, B., Gulcehre, C., Bahdanau, D.,
Bougares, F., Schwenk, H., and Bengio, Y. (2014).
Learning phrase representations using RNN encoder-
decoder for statistical machine translation. arXiv
preprint arXiv:1406.1078.
Corso, G., Ying, Z., P
´
andy, M., Veli
ˇ
ckovi
´
c, P., Leskovec,
J., and Li
`
o, P. (2021). Neural distance embeddings for
biological sequences. Advances in Neural Information
Processing Systems, 34:18539–18551.
Dwivedi, V. P., Ramp
´
a
ˇ
sek, L., Galkin, M., Parviz, A., Wolf,
G., Luu, A. T., and Beaini, D. (2022). Long range
graph benchmark. Advances in Neural Information
Processing Systems, 35:22326–22340.
Fey, M. and Lenssen, J. E. (2019). Fast Graph Represen-
tation Learning with PyTorch Geometric. In ICLR
Workshop on Representation Learning on Graphs and
Manifolds.
Fox, N. K., Brenner, S. E., and Chandonia, J.-M.
(2014). SCOPe: Structural Classification of Pro-
teins—extended, integrating SCOP and ASTRAL
data and classification of new structures. Nucleic acids
research, 42(D1):D304–D309.
Fukushima, K. (1980). Neocognitron: A self-organizing
neural network model for a mechanism of pattern
recognition unaffected by shift in position. Biologi-
cal cybernetics, 36(4):193–202.
Gherardini, P. F. and Helmer-Citterich, M. (2008).
Structure-based function prediction: approaches and
applications. Briefings in Functional Genomics and
Proteomics, 7(4):291–302.
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O., and
Dahl, G. E. (2017). Neural message passing for quan-
tum chemistry. In International conference on ma-
chine learning, pages 1263–1272. PMLR.
Hamilton, W., Ying, Z., and Leskovec, J. (2017). Inductive
representation learning on large graphs. Advances in
neural information processing systems, 30.
Integrating Structure and Sequence: Protein Graph Embeddings via GNNs and LLMs
589

Heinzinger, M., Elnaggar, A., Wang, Y., Dallago, C.,
Nechaev, D., Matthes, F., and Rost, B. (2019). Mod-
eling aspects of the language of life through transfer-
learning protein sequences. BMC bioinformatics,
20(1):1–17.
Henikoff, S. and Henikoff, J. G. (1992). Amino acid sub-
stitution matrices from protein blocks. Proceedings
of the National Academy of Sciences, 89(22):10915–
10919.
Holm, L. and Sander, C. (1993). Protein structure com-
parison by alignment of distance matrices. Journal of
molecular biology, 233(1):123–138.
Holm, L. and Sander, C. (1999). Using dali for structural
comparison of proteins. Current opinion in structural
biology, 9(3):408–415.
Hu, J. X., Thomas, C. E., and Brunak, S. (2016). Network
biology concepts in complex disease comorbidities.
Nature Reviews Genetics, 17(10):615–629.
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov,
M., Ronneberger, O., Tunyasuvunakool, K., Bates, R.,
ˇ
Z
´
ıdek, A., Potapenko, A., et al. (2021). Highly accu-
rate protein structure prediction with AlphaFold. Na-
ture, 596(7873):583–589.
Kihara, D. and Skolnick, J. (2003). The PDB is a covering
set of small protein structures. Journal of molecular
biology, 334(4):793–802.
Kingma, D. P. and Ba, J. (2014). Adam: A
method for stochastic optimization. arXiv preprint
arXiv:1412.6980.
Kipf, T. N. and Welling, M. (2016). Semi-supervised clas-
sification with graph convolutional networks. arXiv
preprint arXiv:1609.02907.
Krishna, S. S., Majumdar, I., Grishin, N., Standley, D., Ru-
binson, E., Wei, L., and Rost, B. (1997). The PDB is
a covering set of small protein structures. Journal of
molecular biology, 267(3):638–657.
Kryshtafovych, A., Schwede, T., Topf, M., Fidelis, K.,
and Moult, J. (2019). Critical assessment of meth-
ods of protein structure prediction (CASP)—Round
XIII. Proteins: Structure, Function, and Bioinformat-
ics, 87(12):1011–1020.
Lathrop, R. H. (1994). The protein threading problem with
sequence amino acid interaction preferences is NP-
complete. Protein Engineering, Design and Selection,
7(9):1059–1068.
Lensink, M. F., Velankar, S., Baek, M., Heo, L., Seok, C.,
and Wodak, S. J. (2018). The challenge of model-
ing protein assemblies: the CASP12-CAPRI experi-
ment. Proteins: Structure, Function, and Bioinfor-
matics, 86:257–273.
Liu, Y., Ye, Q., Wang, L., and Peng, J. (2018). Learning
structural motif representations for efficient protein
structure search. Bioinformatics, 34(17):i773–i780.
Meiler, J., M
¨
uller, M., Zeidler, A., and Schm
¨
aschke, F.
(2001). Generation and evaluation of dimension-
reduced amino acid parameter representations by ar-
tificial neural networks. Molecular modeling annual,
7(9):360–369.
Moreau, Y. and Tranchevent, L.-C. (2012). Computa-
tional tools for prioritizing candidate genes: boost-
ing disease gene discovery. Nature Reviews Genetics,
13(8):523–536.
Morris, R., Black, K. A., and Stollar, E. J. (2022). Uncover-
ing protein function: from classification to complexes.
Essays in Biochemistry, 66(3):255–285.
Nair, V. and Hinton, G. E. (2010). Rectified linear units im-
prove restricted boltzmann machines. In International
conference on machine learning, pages 807–814.
Needleman, S. B. and Wunsch, C. D. (1970). A gen-
eral method applicable to the search for similarities
in the amino acid sequence of two proteins. Journal
of molecular biology, 48(3):443–453.
Røgen, P. and Fain, B. (2003). Automatic classifica-
tion of protein structure by using Gauss integrals.
Proceedings of the National Academy of Sciences,
100(1):119–124.
Scarselli, F., Gori, M., Tsoi, A. C., Hagenbuchner, M., and
Monfardini, G. (2008). The graph neural network
model. IEEE Trans. on neural networks, 20(1):61–80.
Shindyalov, I. N. and Bourne, P. E. (1998). Protein struc-
ture alignment by incremental combinatorial exten-
sion (CE) of the optimal path. Protein engineering,
11(9):739–747.
Srivastava, N., Hinton, G., Krizhevsky, A., Sutskever, I.,
and Salakhutdinov, R. (2014). Dropout: a simple way
to prevent neural networks from overfitting. The jour-
nal of machine learning research, 15(1):1929–1958.
Sterling, T. and Irwin, J. J. (2015). ZINC 15 – ligand dis-
covery for everyone. Journal of Chemical Information
and Modeling, 55(11):2324–2337.
Van der Maaten, L. and Hinton, G. (2008). Visualizing data
using t-SNE. Journal of machine learning research,
9(11).
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones,
L., Gomez, A. N., Kaiser, Ł., and Polosukhin, I.
(2017). Attention is all you need. Advances in neural
information processing systems, 30.
Veli
ˇ
ckovi
´
c, P., Cucurull, G., Casanova, A., Romero, A., Lio,
P., and Bengio, Y. (2017). Graph attention networks.
arXiv preprint arXiv:1710.10903.
Wang, L., Li, Y., and Lazebnik, S. (2016). Learning deep
structure-preserving image-text embeddings. In Pro-
ceedings of the IEEE Conference on Computer Vision
and Pattern Recognition, pages 5005–5013.
Xia, C., Feng, S.-H., Xia, Y., Pan, X., and Shen,
H.-B. (2022). Fast protein structure comparison
through effective representation learning with con-
trastive graph neural networks. PLoS computational
biology, 18(3):e1009986.
Yang, F., Fan, K., Song, D., and Lin, H. (2020). Graph-
based prediction of Protein-protein interactions with
attributed signed graph embedding. BMC bioinfor-
matics, 21(1):1–16.
Zaheer, M., Kottur, S., Ravanbakhsh, S., Poczos, B.,
Salakhutdinov, R. R., and Smola, A. J. (2017). Deep
sets. Advances in neural information processing sys-
tems, 30.
Zhang, C. and DeLisi, C. (1997). A unified statistical frame-
work for sequence comparison and structure compar-
ICPRAM 2024 - 13th International Conference on Pattern Recognition Applications and Methods
590

ison. Proceedings of the National Academy of Sci-
ences, 94(11):5917–5922.
Zhang, Y. and Skolnick, J. (2004). Scoring function for au-
tomated assessment of protein structure template qual-
ity. Proteins: Structure, Function, and Bioinformat-
ics, 57(4):702–710.
Zhang, Y. and Skolnick, J. (2005). TM-align: a protein
structure alignment algorithm based on the TM-score.
Nucleic acids research, 33(7):2302–2309.
Zhou, H., Beltr
´
an, J. F., and Brito, I. L. (2021). Func-
tions predict horizontal gene transfer and the emer-
gence of antibiotic resistance. Science Advances,
7(43):eabj5056.
Zotenko, E., O’Leary, D. P., and Przytycka, T. M. (2006).
Secondary structure spatial conformation footprint: a
novel method for fast protein structure comparison
and classification. BMC Structural Biology, 6:1–12.
APPENDIX
A. GRAPH ARCHITECTURES
A.1 Graph Neural Networks
Graph Neural Networks (GNNs) are a class of neu-
ral networks that operate on data defined over graphs.
Since their introduction (Scarselli et al., 2008), GNNs
have shown outstanding results in a broad range of
applications, from computational chemistry (Gilmer
et al., 2017) to protein folding (Jumper et al., 2021).
The key idea is to exploit the inductive bias induced
by the topology of graph-structured data to perform
graph representation learning tasks.
A graph G = (V, E) is a structure that consists of a
set V of n nodes and a set of edges E. In this context,
each node v ∈ V is equipped with a d-dimensional fea-
ture vector x
v
, and these can be grouped into a feature
matrix X ∈ R
n×d
by stacking all the n = |V | feature
vectors vertically. The connectivity structure of G is
fully captured by the adjacency matrix A, in which the
entry i, j of A is equal to 1 if node i is connected to
node j and 0 otherwise. In GNNs, each layer consists
of a nonlinear function that maps a feature matrix into
a new (hidden) feature matrix, accounting for the pair-
wise relationships in the underlying graph captured by
its connectivity. Formally,
H
(l)
= f (H
(l−1)
;A) (2)
where H
(l)
is the hidden feature matrix at layer l and
H
(0)
= X. Among the plethora of neural architectures
that have this structure, one of the most popular is
the Graph Convolutional Network Kipf and Welling
(2016), which implements Equation 2 as
H
(l)
= σ(
˜
D
−
1
2
˜
A
˜
D
−
1
2
H
(l−1)
W
(l)
) (3)
where W
(l)
is a learnable weight matrix,
˜
A = A + I,
˜
D is a diagonal matrix whose entries are
˜
D
ii
=
∑
j
˜
A
i j
and σ is a pointwise nonlinear activation function (for
example, Sigmoid, Tanh, ReLU).
A.2 Graph Attention Network
The Graph Attention Network (GAT) Veli
ˇ
ckovi
´
c et al.
(2017) is a type of GNN that uses attention mech-
anisms to capture dependencies between nodes in a
graph. The key idea behind GATs is to learn a differ-
ent weight for each neighboring node in the graph us-
ing a shared attention mechanism. This allows a GAT
to attend to different parts of the graph when comput-
ing the representation of each node. The GAT layer
can be mathematically expressed as
h
(l+1)
i
= σ
∑
j∈N (i)
α
(l)
i j
W
(l)
h
(l)
j
(4)
where h
(l)
i
denotes the representation of node i at layer
l, N (i) represents the set of neighbouring nodes of i,
α
(l)
i j
represents the attention weight between nodes i
and j at layer l, W
(l)
is the weight matrix at layer l,
and σ is the activation function. The coefficients com-
puted by the attention mechanism can be expressed
as:
α
i j
=
exp
LeakyReLU
a
⊤
[W
(l)
h
(l)
i
||W
(l)
h
(l)
j
]
∑
k∈N (i)
exp
LeakyReLU
a
⊤
[W
(l)
h
(l)
i
||W
(l)
h
(l)
k
]
(5)
where [·||·] denotes concatenation, a
⊤
is a train-
able weight vector, and LeakyReLU is the Leaky Rec-
tified Linear Unit activation function.
A.3 GraphSAGE
GraphSAGE (Hamilton et al., 2017) is a type of GNN
that learns node representations by aggregating infor-
mation from the local neighborhood of each node.
GraphSAGE learns a set of functions to aggregate
the representations of a node’s neighbors, and then
combine them with the node’s own representation to
compute its updated representation. The GraphSAGE
layer can be mathematically expressed as
h
(l+1)
i
= σ
W
(l)
· CAT
AGG
h
(l)
j
: j ∈ N (i)
, h
(l)
i
(6)
where h
(l)
i
denotes the representation of node i
at layer l, N (i) represents the set of neighbouring
nodes of i, AGG is a learnable aggregation function
that combines the representations of a node’s neigh-
bors, CAT is the concatenation operation, W
(l)
is the
weight matrix at layer l, and σ is the activation func-
tion.
Integrating Structure and Sequence: Protein Graph Embeddings via GNNs and LLMs
591

B. DISTANCE FUNCTIONS
The proposed approach is to map graphs into a con-
tinuous space so that the distance between embedded
points is correlated to the distance between the orig-
inal graphs measured by the TM-score. We explored
different distance functions in the embedding space,
and we give here their definitions. Given a pair of
vectors p and q of dimension k, the definitions of the
Manhattan, Euclidean, Square and Cosine distances
are as follows:
Manhattan: d(p, q) = ∥p − q∥
1
=
k
∑
i=0
|p
i
− q
i
|
Euclidean: d(p, q) = ∥p − q∥
2
=
v
u
u
t
k
∑
i=0
(p
i
− q
i
)
2
Square: d(p, q) = ∥p − q∥
2
2
=
k
∑
i=0
(p
i
− q
i
)
2
Cosine: d(p, q) = 1 −
p·q
∥p∥∥q∥
= 1 −
∑
k
i=0
p
i
q
i
q
∑
k
i=0
p
2
i
q
∑
k
i=0
q
2
i
C. DATASETS
C1. Kinase Proteins
We downloaded the human proteome from UniProt
5
and sub-selected 512 protein kinases. We also used
UniProt to download the PDB files for the kinases.
C2. SCOPe v2.07
The 40% identity filtered subset of SCOPe v2.07
6
is
used to train and validate our approach. Out of the to-
tal of 14,323 domains, 1,058 domains were removed
during the data collection process. The remaining
13,265 domains were used for training and testing.
For both datasets, we computed ground truth TM-
scores by performing all-against-all comparisons us-
ing TM-align (Zhang and Skolnick, 2005). We used
80% of the comparisons for training and 20% for test-
ing. We repeated all the experiments with 3 different
seeds.
5
https://www.uniprot.org
6
https://scop.berkeley.edu/help/ver=2.07
D. ADDITIONAL EXPERIMENTS
AND DETAILS
D1. TM-Scores Predictions
We employed the trained GAT architectures from Ta-
ble 2 to predict the TM-scores for the kinase pairs in
the test set. Results of this evaluation, measured by
Pearson correlation between model predictions and
true TM-scores, are shown in Table 5.
Table 5: Pearson correlation coefficients between predicted
and actual TM-scores for the GAT model for different
choices of node features and distance functions. We use
gold , silver , and bronze colors to indicate the first,
second and third best performances, respectively. The high-
est score is reached with LSTM-extracted features and Eu-
clidean geometry.
Feature Distance
Cosine Euclidean Manhattan Square
One-Hot 0.661 0.384 0.637 0.226
Physicochemical 0.463 0.358 0.534 0.166
BLOSUM 0.484 0.658 0.761 0.468
BERT features 0.849 0.870 0.837 0.785
LSTM features 0.861 0.879 0.858 0.839
Using features learned by LLMs exhibits supe-
rior performance compared to other feature extraction
methods. The highest score is reached with LSTM-
extracted features and Euclidean geometry of the em-
bedding space.
D2. TM-Scores Inference Times
Table 6: Wall-clock estimates for the GNN models and TM-
align on different percentages of the test set. Among the
GNNs, GAT is the slowest at computing TM-scores, fol-
lowed by GraphSAGE and GCN, both on GPU and CPU.
However, TM-score computation with any of the GNN ar-
chitectures is significantly faster than TM-align, even on
CPU.
Test Size (%) Model GPU Inference (s) CPU Inference (s)
26164 (20%)
GCN 88.3 ± 2.04 474.78 ± 1.98
GAT 125.8 ± 2.26 1570.1 ± 3.21
GraphSAGE 98.2 ± 3.46 618.2 ± 2.56
TM-align - 57659.3 ≈ 16 hr
13082 (10%)
GCN 49.2 ± 0.53 231.1 ± 3.01
GAT 59.3 ± 2.34 773.6 ± 3.14
GraphSAGE 49.3 ± 0.04 313.9 ± 2.23
TM-align - 29156.2 ≈ 8 hr
6541 (5%)
GCN 23.2 ± 0.18 119.6 ± 1.76
GAT 30.1 ± 0.70 3882 ± 3.26
GraphSAGE 25.6 ± 1.42 153.1 ± 3.01
TM-align - 15019.9 ≈ 4 hr
Table 6 provides inference times for the different
graph models and TM-align. We show the inference
times on GPU and CPU for the graph models, and
ICPRAM 2024 - 13th International Conference on Pattern Recognition Applications and Methods
592

CPU time for TM-align. Time estimates for different
percentages of the test set (20%, 10%, 5%) are re-
ported. For the graph models, we also report standard
deviations by estimating the times over 5 different
runs. The GNN architectures are significantly faster
than TM-align, even on CPU. Our approach repre-
sents a fast (Table 6) and accurate (Table 5) way to
compute protein structural similarities even on large-
scale datasets.
D3. MSE Results on SCOPe Proteins
Table 7: MSE scores for different distance functions and
LSTM features on the SCOPe dataset. We use gold , silver
, and bronze colors to indicate the first, second and third
best performances, respectively.
Model Distance MSE
GAT
Cosine 0.008048
Euclidean 0.006294
Manhattan 0.010655
Square 0.008793
Table 7 reports the MSE scores for different distance
functions and LSTM features on the SCOPe dataset.
The best MSE is again reached with LSTM-extracted
features and Euclidean geometry of the embedding
space.
D4. Computational Resources, Code
Assets and Data Availability
In all experiments we used NVIDIA
®
Tesla V100
GPUs with 5,120 CUDA cores and 32GB GPU mem-
ory on a personal computing platform with an Intel
®
Xeon
®
Gold 5218 CPU @ 2.30GHz CPU running
Ubuntu 18.04.6 LTS. Our code and the datasets used
for evaluations are available on GitHub
7
.
E. BIOINFORMATICS
APPLICATIONS
There are several areas of bioinformatics research
where structural representation of proteins finds use-
ful applications. We now give a few examples.
E1. Protein-Protein Interaction
Proteins rarely carry out their tasks in isolation, but
interact with other proteins present in their surround-
7
https://github.com/cecca46/neural embeddings
ings to complete biological activities. Knowledge of
protein–protein interactions (PPIs) helps unravel cel-
lular behaviour and functionality. Generating mean-
ingful representations of proteins based on chemical
and structural information to predict protein-pocket
ligand interactions and protein-protein interactions is
an essential bioinformatics task (Yang et al., 2020).
E2. Protein Function
The structural features of a protein determine a wide
range of functions: from binding specificity and con-
ferring of mechanical stability, to catalysis of bio-
chemical reactions, transport, and signal transduc-
tion. While the experimental characterization of a
protein’s functionality is a challenging and intense
task (Moreau and Tranchevent, 2012), exploiting
graph representation learning ability to incorporate
structural information facilitates the prediction of pro-
tein function (Zhou et al., 2021).
E3. Small Molecules
The design of a new drug requires experimentalists to
identify the chemical structure of the candidate drug,
its target, its efficacy and toxicity and its potential side
effects (Hu et al., 2016). Because such processes are
costly and time consuming, drug-discovery pipelines
employ in silico approaches. Effective representa-
tions of protein targets of small molecules (drugs) has
the potential to dramatically speed up the field of drug
discovery.
Integrating Structure and Sequence: Protein Graph Embeddings via GNNs and LLMs
593
