
Assembling Close Strains in Metagenome Assemblies Using Discrete
Optimization
Tam Khac Minh Truong
1,∗
, Roland Faure
1,2,∗ a
and Rumen Andonov
1 b
1
Univ. Rennes, INRIA RBA, CNRS UMR 6074, Rennes, France
2
Service Evolution Biologique et Ecologie, Universit
´
e Libre de Bruxelles (ULB), 1050 Brussels, Belgium
Keywords:
Metagenomes, Strain Separation, Integer Linear Programming.
Abstract:
Metagenomic assembly is essential for understanding microbial communities but faces challenges in distin-
guishing conspecific bacterial strains. This is especially true when dealing with low-accuracy sequencing reads
such as PacBio CLR and Oxford Nanopore. While these technologies provide unequaled throughput and read
length, the high error rate makes it difficult to distinguish close bacterial strains. Consequently, current de novo
metagenome assembly methods excel to assemble dominant species but struggle to reconstruct low-abundance
strains. In our study, we innovate by approaching strain separation as an Integer Linear Programming (ILP)
problem. We introduce a strain-separation module, strainMiner, and integrate it into an established pipeline
to create strain-separated assemblies from sequencing data. Across simulated and real experiments encom-
passing a wide range of error rates (5-12%), our tool consistently compared favorably to the state-of-the-art in
terms of assembly quality and strain reconstruction. Moreover, strainMiner substantially cuts down the com-
putational burden of strain-level assembly compared to published software by leveraging the powerful Gurobi
solver. We think the new methodological ideas presented in this paper will help democratizing strain-separated
assembly.
1 INTRODUCTION
In the field of metagenomic sequencing, the analysis
of bacterial communities is a complex undertaking,
complicated by the presence of conspecific strains
(i.e. strains of the same species). Current de novo
metagenome assembly methods can reconstruct the
chromosomal sequences of prevalent species but gen-
erally struggle to produce strain-level reconstructions.
This capability is vital for discerning subtle genetic
differences among microorganisms that hold crucial
functional significance in different environments. For
instance, many Escherichia coli strains are commen-
sal, while others are pathogenic (Frank et al., 2011).
The challenge posed by the “strain separation
problem,” as outlined in (Vicedomini et al., 2021),
arises from two primary factors: the uncertain and
potentially substantial quantity of conspecific strains
and their uneven distribution within the sample. In
this study, we will use the term “haplotype” to de-
a
https://orcid.org/0000-0003-2245-4284
b
https://orcid.org/0000-0003-4842-7102
∗
These authors contributed equally to this work.
scribe each strain’s genome. It is important to note
that this problem isn’t perfectly defined because the
concept of a “strain” lacks absolute clarity, given that
any two individuals naturally exhibit some genetic
distinctions. Within the scope of this research, we
consider a haplotype to be a contiguous sequence of
nucleotides present in sufficient abundance within the
sample. This approach aligns with similar practices
found in other relevant works, including (Vicedomini
et al., 2021).
Numerous studies have focused on addressing the
strain separation problem primarily in the context
of short-read sequencing. Methods like DESMAN
(Quince et al., 2017), STRONG (Quince et al., 2020),
ConStrains, LSA (Cleary et al., 2015), OPERA-MS
(Bertrand et al., 2019), SAVAGE (Baaijens et al.,
2017), and strainXpress (Kang et al., 2022) have
made notable attempts to tackle this problem, albeit
with various limitations. Additionally, StrainPhlAn
(Truong et al., 2017) and StrainEst (Albanese and
Donati, 2017) have introduced references-based tech-
niques to profile communities and finely distinguish
known strains.
However, with the advent of long-read metage-
Truong, T., Faure, R. and Andonov, R.
Assembling Close Strains in Metagenome Assemblies Using Discrete Optimization.
DOI: 10.5220/0012320700003657
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 17th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2024) - Volume 1, pages 347-356
ISBN: 978-989-758-688-0; ISSN: 2184-4305
Proceedings Copyright © 2024 by SCITEPRESS – Science and Technology Publications, Lda.
347

nomic sequencing, previous methods designed for
short-read data are no longer suitable due to funda-
mental differences in data characteristics. Existing
long-read assemblers, including Canu (Koren et al.,
2017) and metaFlye (Kolmogorov et al., 2020), have
emerged as state-of-the-art solutions for metagenome
assembly but are not designed to fully address the
strain separation challenge.
PacBio High-Fidelity (HiFi) sequencing technol-
ogy has emerged as a promising solution for metage-
nomic strain separation, as the extremely low error
rate can help distinguish very similar sequences. Spe-
cialized software such as hifiasm-meta (Feng et al.,
2022), strainFlye (Fedarko et al., 2022) and stRainy
(Kazantseva et al., 2023) have been developed to
exploit this kind of data. However, achieving suf-
ficient coverage to recover rare strains in complex
metagenomes is challenging and expensive. In this ar-
ticle we will focus on higher-error rates technologies
such as Oxford Nanopore and Pacbio CLR, which are
more affordable and have higher throughput.
MagPhase (Bickhart et al., 2022) and iGDA (Feng
et al., 2021) have been proposed to phase Single
Nucleotide Polymorphisms (SNPs) in metagenomes.
However, these tools were not meant to recover
full haplotypes, but rather phase specific regions of
genomes. MagPhase only returns lists of SNPs, ne-
glecting more complex variants, while iGDA failed to
run on the full metagenomes on which we tested it.
Strainberry (Vicedomini et al., 2021) has been the
first tool to propose a solution to tackle the strain
separation problem at the scale of full metagenome
assembly. It applies iteratively HapCUT2 (Bansal,
2022), a tool developed for diploid phasing. The au-
thors proved that Strainberry was capable of phasing
simple genomes, but the tool is intrinsically limited
to simple metagenomes, i.e. no more two or three
conspecific strains. More recently, the software Hair-
Splitter (Faure et al., 2023) has been introduced. Hair-
Splitter begins by aligning all the reads on a first draft
assembly and calls polymorphisms. It then clusters
reads by similarity, aiming to obtain one cluster per
strain. Finally, it re-assembles the reads to obtain the
phased assembly.
In this article, we introduce a new method to
separate strains in metagenome assemblies using
error-prone long reads. This method introduces
a completely new methodological tool by framing
strain separation as an Integer Linear Programming
(ILP) problem, allowing us to harness the power
of the Gurobi solver (through gurobipy v.10.0.3,
gurobi.com) to achieve high computational efficiency
(Gurobi Optimization, LLC, 2023). We implement
this method in an algorithm named strainMiner. We
(i) Alignment
Reads
Assembly
(ii) Separation
windows
(iii) Re-assembly
Reassembled
contigs
(iv) Scaolding
New
assembly
Read groups
Figure 1: The strainMiner pipeline: (i) Reads are aligned on
the reference or draft assembly, (ii) on each window of the
assembly, reads are separated by haplotype of origin - only
three windows are shown here, (iii) all groups of reads are
locally reassembled and (iv) the locally reassembled contigs
are scaffolded to produce longer contigs.
integrate it in the HairSplitter pipeline to obtain a
complete software capable of producing a new assem-
bly, which we will call “the strainMiner pipeline”.
We show that on mock communities (based on real
and simulated data) the strainMiner pipeline improves
over Strainberry and compares favorably with Hair-
Splitter to phase metagenome assemblies in terms
of assembly completeness, while being an order of
magnitude faster (than Strainberry) or more memory-
efficient (than HairSplitter).
2 CONTRIBUTION
2.1 Pipeline
The strainMiner pipelines takes as input a draft as-
sembly or a reference genome and a set of long
reads and aims at producing a strain-separated assem-
bly. It comprises four primary stages and is illus-
trated Figure 1. These stages include: (i) aligning
the reads to the draft assembly or reference genome,
(ii) the strainMiner algorithm - clustering the reads
based on their respective haplotypes, (iii) conducting
haplotype-specific assembly of the reads, and (iv) en-
hancing assembly contiguity through scaffolding. Ex-
cluding step 2, this pipeline mirrors the one detailed
in (Faure et al., 2023).
Our contribution lies in an original method to pro-
pose a solution for the second step of the pipeline,
i.e. separating aligned reads by haplotype of origin.
The rest of the pipeline was forked from (Faure et al.,
2023), replacing a native HairSplitter module with a
module of our own.
The specific challenge we are addressing can be
described as follows: given a set of reads aligned to
a reference sequence, distinguish groups of reads ac-
cording to their haplotype of origin. In an ideal sce-
BIOINFORMATICS 2024 - 15th International Conference on Bioinformatics Models, Methods and Algorithms
348

nario, all the reads of the dataset originating from
the same strain would be grouped together. How-
ever, achieving this level of separation across an en-
tire genome is not always feasible. Indeed, it is im-
possible to phase two consecutive variants when they
are too far apart to be spanned by at least one read.
Therefore, our task focuses on separating reads lo-
cally by their haplotype of origin. Here we choose
to cut the genome in windows of length w, where w
should be smaller than the average read length (5000
by default).
2.2 Intuition
The intuition behind strainMiner is similar to the one
behind (Faure et al., 2023), originally introduced in
(Feng et al., 2021).
The crucial information for segregating reads by
their haplotype of origin is found within the poly-
morphic sites—specific genomic locations where dif-
ferent strains exhibit variations. However, it is im-
possible to distinguish on a given read a sequencing
error from a variation at a polymorphic locus. Us-
ing the pileup of read might give us an indication, if
many reads show the same nucleotide at the same lo-
cus. Nevertheless, this approach has its limitations,
as alignment artifacts can also create such loci in the
pileup, and the signal from rare strains may not be
very pronounced. The trick lies in examining multi-
ple loci simultaneously, as reads originating from a
particular strain will consistently display similar be-
havior at polymorphic sites. Conversely, the base
pileup between two non-polymorphic sites should not
exhibit significant correlation since alignment or se-
quencing errors occur randomly among all the reads.
As a result, strainMiner endeavors to identify a set
of positions which share identical patterns of varia-
tion. We expect these to represent, for example, all
loci bearing variants specific to one strain.
This strategy allows us to detect even rare strains
effectively. For instance, consider a hypothetical sce-
nario involving a mixture of two strains, where strain
A constitutes 99% of the mix and strain B a mere
1%. Consider also a collection of a thousand reads
spanning two polymorphic sites, denoted as a and
b. In an ideal, error-free pileup, conducting a chi-
square test for independence with one degree of free-
dom between the two loci yields a p-value smaller
than 10
−215
.
However, the introduction of errors significantly
diminishes the statistical power. In our simulations,
we introduced random substitution errors with a prob-
ability of p = 0.1 for all bases across 10,000 sim-
ulations. The p-value for the correlation between a
and b remained low, averaging 10
−16
and reaching
10
−6
in the most unfavorable scenario. Neverthe-
less, it is important to consider that thousands of non-
polymorphic positions can potentially correlate with a
in one pileup. We need also to emphasize that align-
ment artifacts can introduce more complex errors with
locally higher error rates, further reducing the statisti-
cal power of correlation.
The solution is to include more loci to drastically
reduce the risk of spurious correlations. In this same
example, we introduced a third locus, denoted as c,
while maintaining the 0.1 error rate. We applied the
one-degree of freedom chi-square test to assess the re-
lationship between the three positions. This time, the
probability of encountering three non-polymorphic
positions with correlations as strong as those observed
between a, b, and c by chance was found to be be-
low 10
−200
in all 10,000 simulations. While this ex-
ample simplifies the complexities of pileup errors, it
emphasizes two fundamental aspects of the method:
a) the joint observation of multiple loci significantly
enhances the statistical power to distinguish between
errors and polymorphism, and b) even low-abundance
strains can be reliably identified.
2.3 Preprocessing
In each window, strainMiner considers only reads that
span at least 60% of the window’s length. Subse-
quently, strainMiner transforms the read pileup into a
binary matrix, where each row corresponds to a read,
each column corresponds to a position, and cell (i, j)
contains the number one if the base at position j of
read i matches the dominant base at that position, the
number zero if it matches the most common alterna-
tive allele, and remains empty otherwise. Empty cells
can occur if a read doesn’t cover a position or if the
base in a read at a given position isn’t among the two
most common bases at that position.
Next, columns are filtered to retain only those in
which the most common base is present in less than
a proportion p of the aligned reads at that position.
By default, strainMiner sets p to 0.95, striking a bal-
ance between computational efficiency and precision.
However, users seeking to ensure the recovery of low-
abundance strains can set p to a higher value.
To populate the empty cells, strainMiner imple-
ments the well-known K-nearest-neighbor imputation
strategy (Fix and Hodges, 1989), used widely, for
example in (Troyanskaya et al., 2001). It identifies
for each read its “nearest neighbors,” i.e. reads with
the smallest hamming-distance proportionally. Then,
for each empty cell in a row, strainMiner uses a ma-
jority vote from the five closest neighbors that have
Assembling Close Strains in Metagenome Assemblies Using Discrete Optimization
349

non-empty cells at that position to impute the missing
value.
The result is a binary matrix filled with zeroes and
ones. Our primary objective is to identify groups of
positions where reads exhibit similar behavior, hence
strainMiner essentially finds sets of similar columns
in the matrix. Each of these groups will represent a
bipartition of strains, distinguishing strains with the
predominant allele from those with the secondary al-
lele.
In practice, this task is considerably more chal-
lenging than it may initially appear. Attempting to
identify position groups through a straightforward hi-
erarchical clustering approach often proves ineffec-
tive. The reason behind this lies in the calculation of
pairwise distances between two positions, which can
be deceptive. In many cases, sequencing and align-
ment errors can overshadow small differences result-
ing from low-abundance strains. As emphasized ear-
lier, the key is to jointly examine more than just two
loci simultaneously.
2.4 Finding Bipartitions of Reads
The approach used to identify bipartitions is depicted
in Figure 2. It begins by identifying the largest bi-
cluster (a cluster of row and columns) of ones in the
matrix, allowing a small number of zeros to be toler-
ated to account for sequencing errors (referred to as
a quasi-bicluster of ones). Figure 2.b visualises the
largest quasi-bicluster found in the first step. Next,
another submatrix is formed, including the columns
from the first quasi-bicluster and all the reads not
included in the initial one. In this new submatrix,
the largest quasi-bicluster of zeros is sought (Fig-
ure 2.c). This process continues, alternating between
quasi-biclusters of ones and zeros, until all reads have
been included in these clusters or all columns ex-
cluded from the biclusters. The columns shared by all
the clusters define a bipartition of reads (Figure 2.e).
These columns are excluded from the total matrix and
the process is repeated to find another bipartition of
reads using the remaining columns.
Once a list of bipartitions has been determined,
reads are split in groups following a very simple ra-
tionale: two reads are in the same group if and only if
they are grouped together in all bipartitions.
2.4.1 Finding Largest Submatrix
In strainMiner, the primary problem is to discover
the largest bicluster during each iteration. We ac-
complish this by employing a Integer Linear Prob-
lem (ILP) model. This model’s purpose is to select
a group of rows and columns where you can find the
highest count of a specific value (either 0s or 1s).
This problem is known as finding a maximum edge
biclique problem and it has been proven to be NP-
complete (Peeters, 2003). In strainMiner, we search
for a maximum edge quasi-biclique that tolerates a
small amount of errors.
In the context of a matrix A ∈ Z
|U |×|V |
2
with coef-
ficients being 0 or 1, and with a set of rows U and a
set of columns V , we use binary variables x
i j
, u
i
and
v
j
to denote cell selection, row selection, and column
selection, respectively. Here, a binary variable equals
1 to indicate the selection of a cell, row, or column,
and 0 otherwise.
The following Integer Linear Program is then for-
mulated:
max
∑
i∈U
∑
j∈V
A
i, j
x
i j
, (1)
x
i j
≤ u
i
, ∀i ∈ U, ∀ j ∈ V (2)
x
i j
≤ v
j
, ∀i ∈ U, ∀ j ∈ V (3)
x
i j
≥ u
i
+ v
j
− 1, ∀i ∈ U, ∀ j ∈ V (4)
∑
i∈U
∑
j∈V
(1 − A
i, j
)x
i j
≤ ε ×
∑
i∈U
∑
j∈V
x
i j
(5)
u
i
, v
j
∈ {0, 1}, x
i j
∈ {0, 1} ∀i ∈ U, ∀ j ∈ V (6)
The function to maximize, (1), counts for the
number of ones in a submatrix determined by the bi-
nary variables having value 1. Constraints (2), (3),
(4) mean that cell A
i j
is selected into the solution (i.e.
x
i j
= 1) if and only if its corresponding row i and col-
umn j are also chosen into the solution (i.e. u
i
= 1
and v
j
= 1).
The coefficient A
i j
represents the value of the cell
at position i and j, when searching for occurrences of
1s in the matrix, A
i j
is used directly. However, if the
search is for 0s, the coefficient is reversed to (1−A
i j
):
max
∑
i∈U
∑
j∈V
(1 − A
i, j
)x
i j
(7)
Constraint (5) ensures that the submatrix contains
at least a proportion 1 −ε of ones. This constraint can
be reversed to ensure a minimum proportion of zeros:
∑
i∈U
∑
j∈V
A
i, j
x
i j
≤ ε ×
∑
i∈U
∑
j∈V
x
i j
(8)
To identify a bipartition, strainMiner involves
solving a series of this ILP models using the Gurobi
solver, alternating between looking for ones and ze-
ros. This process is repeated to discover new biparti-
tions until the model identifies a submatrix with less
than 5 rows or 5 columns — a threshold empirically
set to identify a statistically significant signal. When
this condition is met, the algorithm terminates and re-
turns the identified bipartitions.
BIOINFORMATICS 2024 - 15th International Conference on Bioinformatics Models, Methods and Algorithms
350
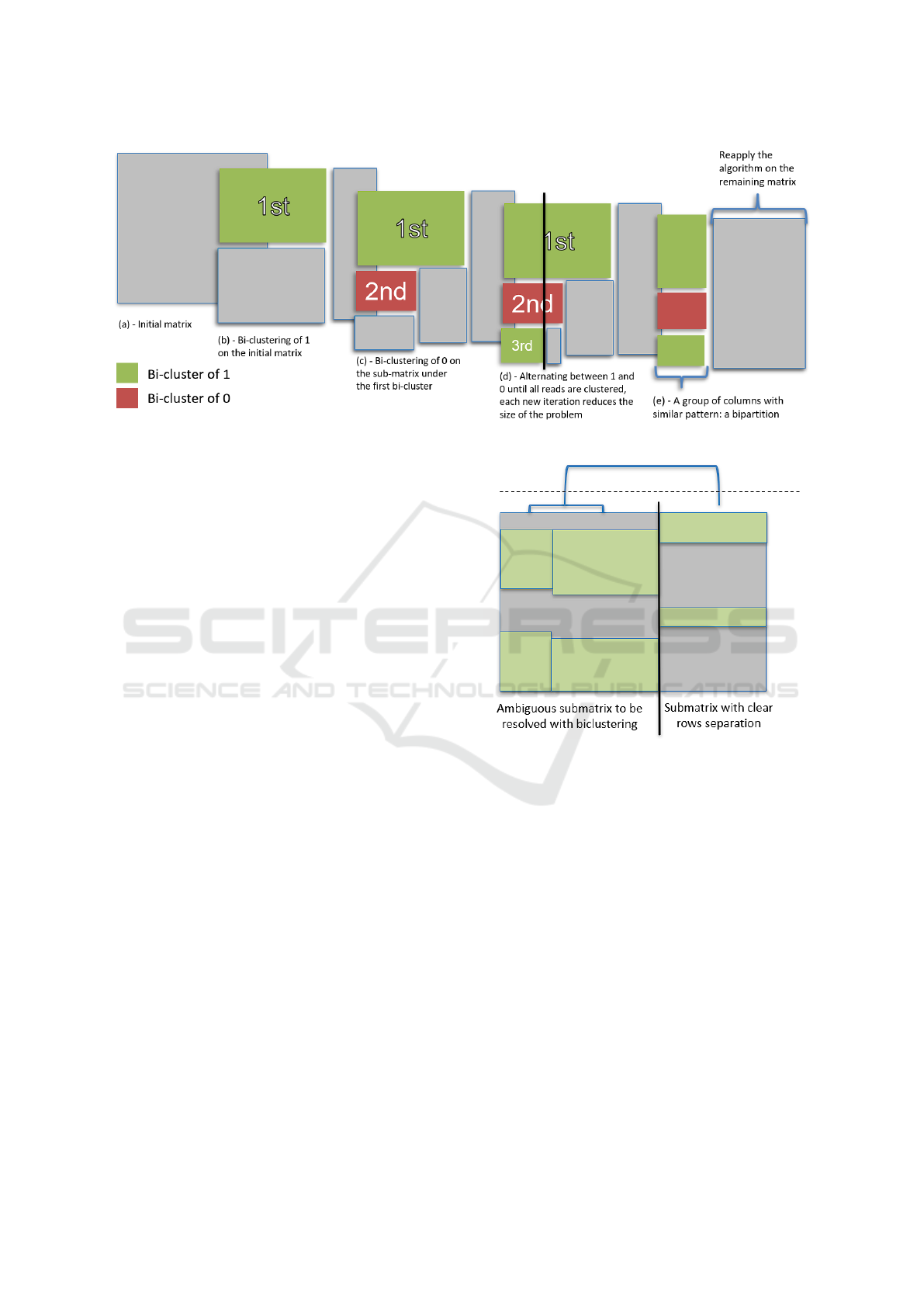
Figure 2: Strategy implemented to find bipartitions.
2.4.2 Acceleration with Divide and Conquer
In order to reduce the computational cost as well as
the runtime of strainMiner, we implement a strategy
aiming to reduce the size of the matrices prior to run-
ning the model.
If a straightforward column-wise hierarchical
clustering cannot be used to make fine distinctions
between bipartitions (for reasons explained above), it
remains an effective strategy for distinguishing highly
divergent positions. Thus, a basic hierarchical cluster-
ing of the positions with Hamming distance and com-
plete linkage is performed. The clusters are formed
by splitting vertically the initial matrix, a threshold of
35% is set as the distance threshold. Clusters with
the linkage distance higher than the limit will not be
merged. This means that two columns in two dif-
ferent clusters have more than 35% divergence and
will never be even considered to be grouped in the
same bipartition - we purposefully chose a conserva-
tive threshold.
The distance threshold needs to balance creating
separate sub-matrices while preserving smaller pat-
terns. If the threshold is too low, even minor data
variations and errors can wrongly divide related posi-
tions, breaking patterns. If it is too high, sub-matrices
that could be split get grouped, forming fewer, larger
sub-matrices and the computational gain is not opti-
mal.
The resulting sub-matrices are subsequently as-
sessed for “ambiguity”. A submatrix consisting of
full rows of zeros or ones is by itself a bipartition
c.f. Figure 3 right. The other submatrices are “am-
biguous” c.f. Figure 3 left. Bipartitions are sought
only in the ambiguous matrices, using the algorithm
described above.
Figure 3: Hierarchical clustering is performed to cut the
matrix into smaller distinct sub-matrices definining distinct
bipartitions.
3 RESULTS
3.1 Datasets
We benchmarked strainMiner on three datasets of
increasing complexity. The first one is a mix of
five Vagococcus fluvialis strains sequenced in (Ro-
driguez Jimenez et al., 2022). The bacteria were
sequenced using a R9.4.1 Nanopore flowcell. The
reads were barcoded by strain of origin, allowing
us to assemble the five strains separately. By ig-
noring the barcodes, we obtain our dataset of five
mixed strains of approximately equal abundance se-
quenced with Nanopore. Three of these strains were
almost identical, approximately turning the problem
in a three-strain separation problem with one predom-
Assembling Close Strains in Metagenome Assemblies Using Discrete Optimization
351
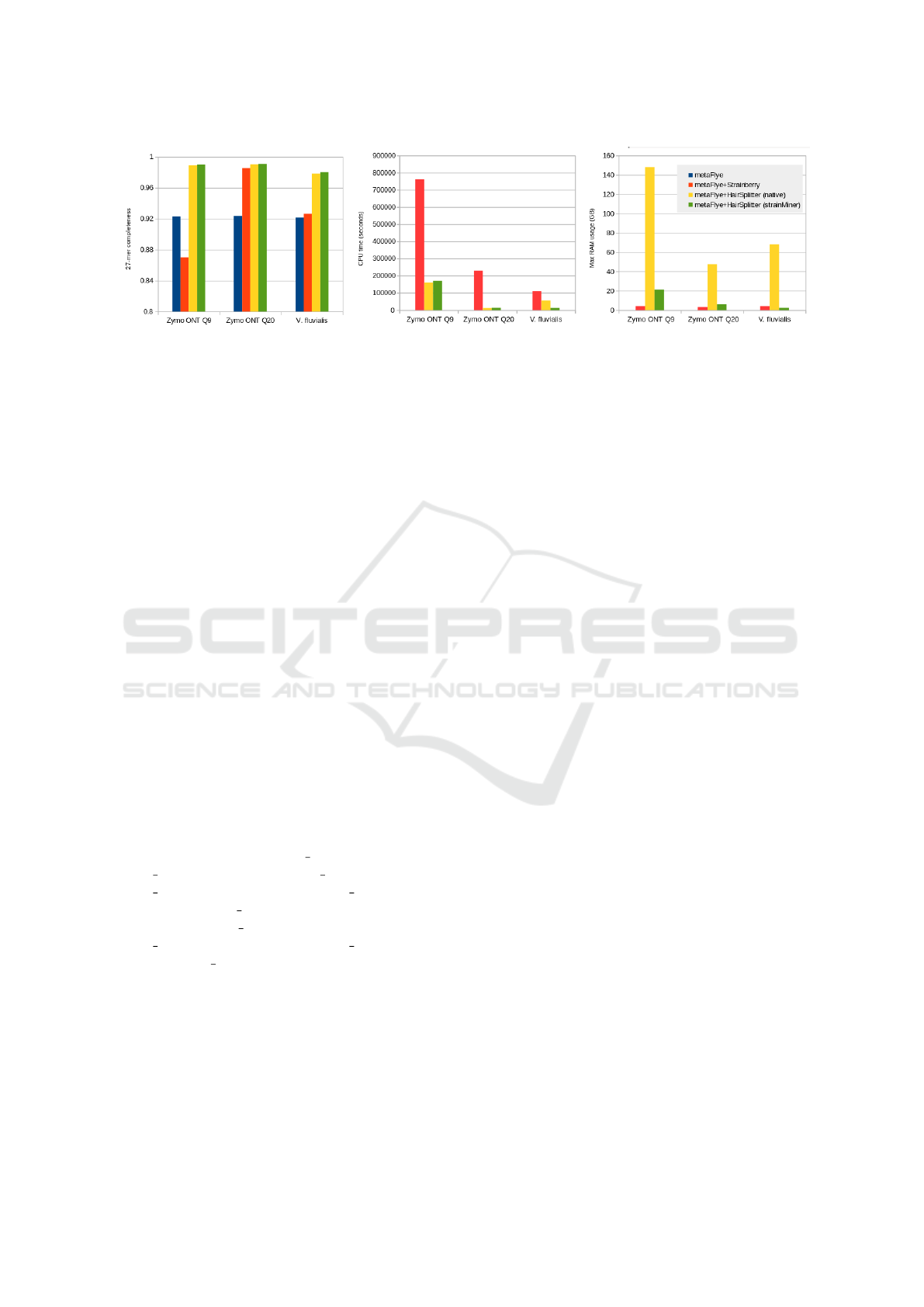
Figure 4: Comparison of Strainberry, HairSplitter and strainMiner on the three real sequencing experiments in terms of, from
left to right, completeness of assembly, CPU time and maximum memory usage. The legend applies to the three plots.
inant species.
The second dataset is the sequencing of the Zy-
mobiomics gut microbiome standard by Nanopore
R9.4.1 (Q9 reads) and Nanopore 10.4.1 (Q20+
reads) flowcells (accession numbers SRR17913199
and SRR17913200). The mix contains a total of 21
strains of bacteria, archea and yeast. In particular, it
contains five Escherichia coli strains in equal abun-
dance, for which we will compare strain separation
techniques. This is the dataset having a known so-
lution with highest number of conspecific strains we
could find.
In a final step, we conducted simulations to
gauge the limits of strainMiner’s strain separation
capabilities. We adapted the simulation protocol
outlined in (Vicedomini et al., 2021) and (Faure
et al., 2023) to investigate how the number of strains
and the depth of coverage influence the effectiveness
of strain separation. We downloaded reference
genomes for ten distinct strains of Escherichia coli
from the SRA and proceeded to simulate Nanopore
reads with a 5% error rate and 50x coverage using
the default “Nanopore2023” mode of Badreads
(Wick, 2019). These reads were then combined to
emulate metagenomic sequencing scenarios. The ten
strains mirrored those used in (Faure et al., 2023)
and included 12009 (GCA 000010745.1), IAI1
(GCA 000026265.1), F11 (GCA 018734065.1), S88
(GCA 000026285.2), Sakai (GCA 003028755.1),
SE15 (GCA 000010485.1), Shigella
flexneri (GCF 000006925.2), UMN026
(GCA 000026325.2), HS (GCA 000017765.1),
and K12 (GCF 009832885.1). The simulated reads
are available at https://zenodo.org/records/10362565.
To assess the impact of the number of strains on
assembly completeness, we assembled and separated
mixtures of 2, 4, 6, 8, and all 10 strains. Additionally,
to evaluate the influence of coverage on assembly
completeness, we downsampled the 12009 strain to
30x, 20x, 10x, and 5x within the context of the 10-
strain mixture, measuring the 27-mer completeness
of the 12009 strain in the various assemblies.
All of these datasets were assembled using
metaFlye (Kolmogorov et al., 2020) because it is to
our knowledge the only noisy long-read assembler
specialized in metagenome assembly. The metaFlye
assembly was then run through the three software.
3.2 Evaluation Metrics
To evaluate the quality of obtained assemblies we
measured assembly length, N50, misassemblies, mis-
matches and indels, measured by software metaQuast
(Mikheenko et al., 2015). We used options –
unique-mapping and –reuse-combined-alignments in
metaQuast to avoid a sequence (a contig, or part of
it) to be mapped on multiple distinct reference lo-
cations. The evaluation of metagenome assemblies
in presence of highly similar references is however a
challenging task. As metaQuast is based on sequence
alignment, it could suffer from a sub-optimal map-
ping of contigs to the references. For this reason,
we decided to complement metaQuast’s metrics by
computing the k-mer completeness (k=27) with KAT
(Mapleson et al., 2016).
3.3 Benchmark
All results were obtained by running the software on
a server housing 16 Intel Xeon CPUs with four cores
each, running at 2.7GHz. 3.1 TB of RAM was avail-
able.
3.3.1 Assembly Evaluation
The summarized metaQuast metrics in Table 1 show
that the strainMiner pipeline yields highly precise
assemblies, exhibiting fewer misassemblies, mis-
matches, and indels compared to Strainberry or Hair-
Splitter. However, this enhanced precision is balanced
by lower contiguity. To manage the size of Table
1, we haven’t included all the E. coli experiments,
BIOINFORMATICS 2024 - 15th International Conference on Bioinformatics Models, Methods and Algorithms
352
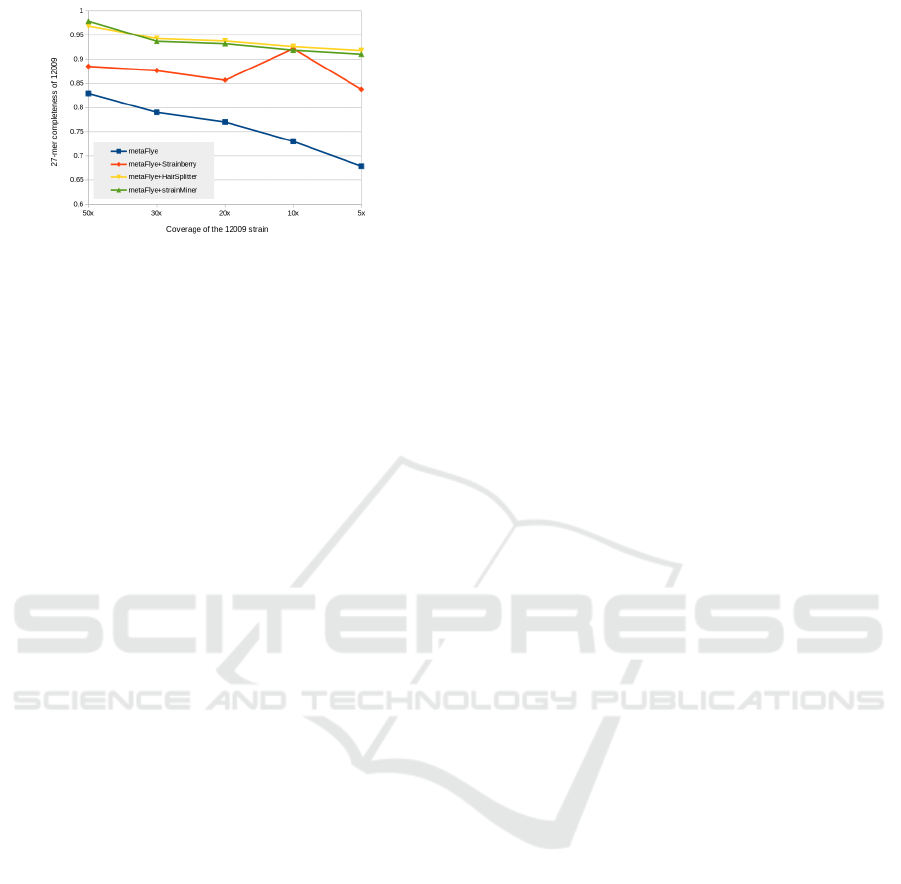
Figure 5: 27-mer completeness of the 12009 strain in a 10-
strain mix. Coverage of the 12009 strain on x axis, the nine
other strains are covered at 50x.
but the downsampling experiments displayed similar
statistics to the 10-strains mix.
On the Zymobiomics and the Vagococcus fluvialis
datasets, the strainMiner pipeline proved the best soft-
ware to recover strain diversity as measured by 27-
mer completeness, though the performances of Hair-
Splitter were almost equivalent (Figure 4). Notably,
the strainMiner pipeline yielded assemblies of equiv-
alent quality when using Q9 and Q20+ chemistry, un-
derscoring its robustness in managing variations in
read quality.
In our experiments with E. coli (Figure 6), strain-
Miner consistently demonstrated its capability to sep-
arate a substantial number of strains, maintaining a
stable 27-mer completeness across scenarios involv-
ing 2, 4, 6, 8, and 10 strains. The strainMiner pipeline
consistently outperformed HairSplitter to a slight ex-
tent in all mix scenarios.
The coverage reduction experiment depicted in
Figure 5 revealed that both strainMiner and Hair-
Splitter exhibited an impressive capability to retrieve
strains with low coverage. Even when the strain’s
coverage dropped to as low as 5x, representing only
1.1% of the total mix, these tools managed to recover
a substantial portion of the previously lost 27-mers.
There remained a noticeable positive correlation be-
tween the coverage level of the downsampled 12009
strain and its resulting completeness.
The performance of Strainberry is difficult to as-
sess, as it performed extremely well in 2-strain, 4-
strain, 8-strain and 20x-downsampling mixtures but
failed to produce good strain-separated assemblies in
the other E. coli scenarios. This inconsistent behav-
ior is probably due to it being conceived for relatively
simple metagenomes (i.e., up to five strains).
These experiments offer insights indicating that
all three software solutions can attain assemblies of
comparable quality, albeit with varying tradeoffs be-
tween contiguity and accuracy, when employing high-
quality Nanopore reads. Figure 4 and additional ex-
aminations performed on mixtures of E. coli with
more error-prone data (not shown) suggest that Strain-
berry encounter difficulties when confronted with
higher error rates.
3.3.2 Resource Usage
All the times reported are total CPU times. Stain-
Miner is trivially parallel, like HairSplitter and Strain-
berry, but in practice we ran it on only one thread be-
cause we were limited by the Gurobi license.
Across all datasets, strainMiner consistently ex-
hibited a processing speed more than tenfold faster
than Strainberry, while its runtime was approximately
on par with that of HairSplitter (Figures 4, 6). For all
three approaches we expect a linear increase in run-
time when increasing the length of the assembly and
the number of species, as aligning is an almost-linear
process in the length of the reference and contigs are
processed independently by the strain-separation soft-
ware.
On the real sequencing data, strainMiner used be-
tween 7 and 30 times less peak memory than Hair-
Splitter (Figure 4).
As a whole, strainMiner significantly diminishes
the memory usage of the HairSplitter pipeline without
impacting negatively on its speed.
4 DISCUSSION
In this manuscript, we introduce strainMiner, an inno-
vative approach aimed at enhancing the performance
of the read-separation module within the HairSplit-
ter strain-separation pipeline, leading to strain-aware
metagenome assemblies. For the first time, we frame
the strain separation as an ILP problem. This al-
lows us to use a well-established, highly optimized
solver to tackle the intensive computations needed
for such a task. By helping the solver with sim-
ple preprocessing techniques inspired from data min-
ing, the final pipeline is considerably more frugal in
time and memory than the state-of-the-art software.
This development presents new opportunities for bi-
ologists, as strain separation is a computationally de-
manding task, and having access to such computa-
tional resources can be a significant constraint for bi-
ology laboratories. On real datasets, the strainMiner
pipeline also compared favorably to published soft-
ware in terms of strain recovery and arguably pro-
vided the most accurate assemblies. Additionally, we
showed that strainMiner could perform well on long
reads with varying error rate without any manual fine-
tuning.
Assembling Close Strains in Metagenome Assemblies Using Discrete Optimization
353

Figure 6: Comparison of Strainberry, HairSplitter and strainMiner on mixes of an increasing number of E. coli strains in terms
of, from left to right, completeness of assembly, CPU time and maximum memory usage. Note that CPU time is represented
in logarithmic scale. The legend applies to the three plots.
Table 1: metaQuast metrics measuring the quality of assemblies on the three real and two simulated datasets. The best value
or best values among of the three strain-separated assemblies (excluding the original assembly) is in bold font.
Total length (Mb) N50 (kb) Misassemblies
Mismatches
/100kbp
Indels
/100kbp
Vagococcus metaFlye 4.35 162 40 340.14 383.57
(14 Mb) Strainberry 5.51 135 135 77.71 510.25
HairSplitter 9.43 82 117 89.93 327.91
strainMiner 8.63 30 48 50.8 340.26
Zymo Q9 metaFlye 61.9 1904 110 91.14 56.28
(78 Mb) Strainberry 62.8 225 122 133.04 109.85
HairSplitter 86.9 74 170 124.54 124.54
strainMiner 68.4 225 95 69.8 55.44
Zymo Q20 metaFlye 49.8 383 126 109.51 75.79
(78 Mb) Strainberry 59.7 121 141 109.12 67.23
HairSplitter 66.2 84 142 75.42 61.23
strainMiner 59.8 51 109 67.97 65.14
E. coli metaFlye 11.6 56 79 469.27 277.71
10 strains Strainberry 18.3 63 175 363.06 137.01
(48 Mb) HairSplitter 50.5 48 812 266.55 68.04
strainMiner 50.1 56 335 81.65 72.73
E. coli metaFlye 6.05 374 22 216.18 216.53
2 strains Strainberry 10.8 1152 11 24.83 64.73
(9.9 Mb) HairSplitter 10.4 230 47 53.75 62.67
strainMiner 9.65 45 8 15.4 59.77
A limitation of the strainMiner pipeline, for now,
is the limited contiguity of the assembly produced.
Adapting the pipeline to the specificities of the strain-
Miner module would be the first lead to pursue to im-
prove this. Indeed, the assemblies obtained through
the strainMiner pipeline had often much lower con-
tiguity compared to the one obtained by HairSplitter,
suggesting that the properties of the read separation
computed by the strainMiner module and the native
HairSplitter read-separation module may be quite dif-
ferent. Tailoring a scaffolding step could thus im-
prove contiguity.
One aspect of strainMiner that would require
further investigation is its approach of dividing the
problem into non-overlapping fixed-length windows.
While this division results in a clean input matrix
for the Integer Linear Programming (ILP) solver, it
introduces an arbitrary separation of loci that may
jointly carry valuable information. This division di-
minishes the sensitivity of strainMiner when dealing
with highly similar strains, as the number of loci in
a single window may be too low for detection, while
there may be enough loci overall to detect the strain.
A possible solution to address this limitation would
be considering overlapping windows. Another option
would be the adaptation of the model to handle miss-
BIOINFORMATICS 2024 - 15th International Conference on Bioinformatics Models, Methods and Algorithms
354

ing values without the need for imputation, which for
now limits the length of the windows.
Finally, users might find themselves limited by the
Gurobi license. Academic license is free but limited
to three instances of Gurobi running at the same time,
limiting the multi-threading potential. Attempts to
use the free CBC solver showed a decrease in per-
formance.
SOFTWARE AVAILABILITY
strainMiner is freely availabe on github at
github.com/RolandFaure/strainMiner, with the
Affero GPL3 license.
ACKNOWLEDGEMENTS
We wish to thank Dominique Lavenier, who formu-
lated the first version of the optimization problem.
Many thanks to Riccardo Vicedomini for his help
in pre-reviewing the article.
ChatGPT was used to correct and reformulate the
writing of the article.
REFERENCES
Albanese, D. and Donati, C. (2017). Strain profiling and
epidemiology of bacterial species from metagenomic
sequencing. Nature Communications, 8.
Baaijens, J., Aabidine, A., Rivals, E., and Sch
¨
onhuth, A.
(2017). De novo assembly of viral quasispecies using
overlap graphs. Genome Research.
Bansal, V. (2022). Hapcut2: A method for phasing genomes
using experimental sequence data. Methods in molec-
ular biology, 2590:139–147.
Bertrand, D., Shaw, J., Kalathiyappan, M., Ng, A. H. Q.,
Kumar, M. S., Li, C., Dvornicic, M., Soldo, J. P.,
Koh, J. Y., Tong, C., Ng, O. T., Barkham, T., Young,
B., Marimuthu, K., Chng, K. R., Sikic, M., and Na-
garajan, N. (2019). Hybrid metagenomic assembly
enables high-resolution analysis of resistance deter-
minants and mobile elements in human microbiomes.
Nature Biotechnology, 37(8):937–944.
Bickhart, D., Kolmogorov, M., Tseng, E., Portik, D., Ko-
robeynikov, A., Tolstoganov, I., Uritskiy, G., Liachko,
I., Sullivan, S., Shin, S., Zorea, A., Pascal, V., Panke-
Buisse, K., Medema, M., Mizrahi, I., Pevzner, P., and
Smith, T. (2022). Generating lineage-resolved, com-
plete metagenome-assembled genomes from complex
microbial communities. Nature Biotechnology, 40.
Cleary, B., Brito, I., Huang, K., Gevers, D., Shea, T.,
Young, S., and Alm, E. (2015). Detection of low-
abundance bacterial strains in metagenomic datasets
by eigengenome partitioning. Nature biotechnology,
33.
Faure, R., Flot, J.-F., and Lavenier, D. (2023). Hairsplit-
ter: separating strains in metagenome assemblies with
long reads. In Proceedings of JOBIM 2023, pages
124–131.
Fedarko, M., Kolmogorov, M., and Pevzner, P. (2022). An-
alyzing rare mutations in metagenomes assembled us-
ing long and accurate reads. Genome research, 32.
Feng, X., Cheng, H., Portik, D., and Li, H. (2022).
Metagenome assembly of high-fidelity long reads
with hifiasm-meta. Nature Methods, 19:1–4.
Feng, Z., Clemente, J., Wong, B., and Schadt, E. (2021).
Detecting and phasing minor single-nucleotide vari-
ants from long-read sequencing data. Nature Commu-
nications, 12:3032.
Fix, E. and Hodges, J. L. (1989). Discriminatory analy-
sis. nonparametric discrimination: Consistency prop-
erties. International Statistical Review / Revue Inter-
nationale de Statistique, 57(3):238–247.
Frank, C., Werber, D., Cramer, J. P., Askar, M., Faber, M.,
an der Heiden, M., Bernard, H., Fruth, A., Prager,
R., Spode, A., Wadl, M., Zoufaly, A., Jordan, S.,
Kemper, M. J., Follin, P., M
¨
uller, L., King, L. A.,
Rosner, B., Buchholz, U., Stark, K., and Krause, G.
(2011). Epidemic profile of shiga-toxin–producing
escherichia coli o104:h4 outbreak in germany. New
England Journal of Medicine, 365(19):1771–1780.
PMID: 21696328.
Gurobi Optimization, LLC (2023). Gurobi [computer soft-
ware]. gurobi.com.
Kang, X., Luo, X., and Sch
¨
onhuth, A. (2022). StrainXpress:
strain aware metagenome assembly from short reads.
Nucleic Acids Research, 50(17):e101–e101.
Kazantseva, E., Donmez, A., Pop, M., and Kolmogorov, M.
(2023). stRainy: assembly-based metagenomic strain
phasing using long reads. preprint, Bioinformatics.
Kolmogorov, M., Bickhart, D. M., Behsaz, B., Gurevich,
A., Rayko, M., Shin, S. B., Kuhn, K., Yuan, J., Pole-
vikov, E., Smith, T. P. L., and Pevzner, P. A. (2020).
metaFlye: scalable long-read metagenome assembly
using repeat graphs. Nature Methods, 17(11):1103–
1110.
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman,
N. H., and Phillippy, A. M. (2017). Canu: scalable
and accurate long-read assembly via adaptive k -mer
weighting and repeat separation. Genome Research,
27(5):722–736.
Mapleson, D., Accinelli, G., Kettleborough, G., Wright, J.,
and Clavijo, B. (2016). Kat: A k-mer analysis toolkit
to quality control ngs datasets and genome assemblies.
Bioinformatics (Oxford, England), 33.
Mikheenko, A., Saveliev, V., and Gurevich, A. (2015).
Metaquast: Evaluation of metagenome assemblies.
Bioinformatics, 32:btv697.
Peeters, R. (2003). The maximum edge biclique problem is
NP-complete. 131:651–654.
Quince, C., Delmont, T. O., Raguideau, S., Alneberg, J.,
Darling, A. E., Collins, G., and Eren, A. M. (2017).
Assembling Close Strains in Metagenome Assemblies Using Discrete Optimization
355

DESMAN: a new tool for de novo extraction of strains
from metagenomes. Genome Biology, 18(1):181.
Quince, C., Nurk, S., Raguideau, S., James, R., Soyer, O. S.,
Summers, J. K., Limasset, A., Eren, A. M., Chikhi,
R., and Darling, A. E. (2020). Metagenomics Strain
Resolution on Assembly Graphs. preprint, Bioinfor-
matics.
Rodriguez Jimenez, A., Guiglielmoni, N., Goetghebuer,
L., Dechamps, E., George, I., and Flot, J.-F. (2022).
Comparative genome analysis of vagococcus fluvi-
alis reveals abundance of mobile genetic elements in
sponge-isolated strains. BMC Genomics, 23.
Troyanskaya, O., Cantor, M., Sherlock, G., Brown, P.,
Hastie, T., Tibshirani, R., Botstein, D., and Altman,
R. B. (2001). Missing value estimation methods for
dna microarrays. Bioinformatics, 17(6):520–525.
Truong, D. T., Tett, A., Pasolli, E., Huttenhower, C., and
Segata, N. (2017). Microbial strain-level population
structure and genetic diversity from metagenomes.
Genome Research, 27:gr.216242.116.
Vicedomini, R., Quince, C., Darling, A. E., and Chikhi, R.
(2021). Strainberry: automated strain separation in
low-complexity metagenomes using long reads. Na-
ture Communications, 12(1):4485.
Wick, R. (2019). Badread: simulation of error-prone long
reads. Journal of Open Source Software, 4(36):1316.
BIOINFORMATICS 2024 - 15th International Conference on Bioinformatics Models, Methods and Algorithms
356
