
The Profile of SARS-Cov-2 Genome from Indonesia and Its Impact
on Paxlovid
TM
in Treating Covid-19
Hotma Martogi Lorensi Hutapea
1,2,* a
, Yustinus Maladan
2b
, Tri Yunis Miko Wahyono
1
,
Arli Aditya Parikesit
3c
and Mondastri Korib Sudaryo
1d
1
Department of Epidemiology, Faculty of Public Health, Universitas Indonesia, Depok, Jawa Barat, Indonesia
2
National Research and Innovation Agency, Jl. M.H. Thamrin No. 8, Jakarta 10340, Indonesia
3
Department of Bioinformatics, School of Life Sciences, Indonesia International Institute for Life Sciences (i3L), Jakarta
13210, Indonesia
Keywords: 3CLPro, Molecular Docking, Nirmatrelvir, Paxlovid
TM
, SARS-CoV-2 Genome.
Abstract: Since April 2023, Paxlovid
TM
has been used to treat SARS-CoV-2 infection in Indonesia. This medication
contains 2 active compounds, ritonavir (RTV) and nirmatrelvir (NTV) that act as human CYP3A4 and viral
main proteinase or 3CL
Pro
respectively. NTV is novel drug to inhibit SARS-CoV-2 progressivity. Mutations
that decrease NTV effectiveness have been reported in several countries, however, the data from Indonesia is
still limited. We are using SARS-CoV-2 genomic data from GISAID which were collected from Sept 1, 2022
– Oct 31, 2023, from Indonesia. Any mutation in 3CL
Pro
was recorded and then proceeded to be analysed
further. NTV-3CL
Pro
complex was downloaded from the Protein data bank (7SI9), and separated in PDB
format. The 3CL
Pro
was mutated using FOLDX to generate the mutant variant. Docking simulation was
performed using Autodock Vina which is integrated into PyRx.0.9.7 software with SARS-CoV-2 Wuhan-Hu
isolate (NC_045512.2) as control. RMSD score was calculated using YASARA software and considered valid
if the redocking score is <2.0 Å. In total, 13,345 genomes from COVID-19 patients were submitted in GISAID,
and all of them were Omicron, with GRA clade and XBB subvariant being more predominant. We found out
that 3CL
Pro
encoding genes were relatively conserved with only P132H mutation motive (99.8%) identified.
Therefore, we performed the simulation on mutant P184L which was found in the Delta variant. Docking
simulation demonstrated that the binding affinity score between nirmatrelvir and control was -8.6 kcal/mol,
and -8.5 kcal/mol on 3CL
Pro
mutant. According to the visualization of NTV-3CL
Pro
interaction, the mutant
P132 and P184 showed no difference compared to the control. We found no mutation that potentially
decreased the effectiveness of Paxlovid
TM
based on the activity of NTV in the SARS-CoV-2 genome collected
from patients in Indonesia. Therefore, SARS-CoV-2 in Indonesia might be susceptible to Paxlovid
TM
.
1 INTRODUCTION
SARS-CoV-2 infection causes coronavirus disease
2019 (COVID-19), which has caused approximately
6.7 million cases and over 160 thousand fatalities in
Indonesia as of March 2023 (World Health
Organization, 2023b). It remains a major public
health threat globally and has been indicated that the
virus will most likely remain as an established
a
https://orcid.org/0000-0002-7099-3891
b
https://orcid.org/0000-0002-6685-7790
c
https://orcid.org/0000-0001-8716-3926
d
https://orcid.org/0000-0003-0896-1538
pathogen in humans and animals for a long time. On
April 2023, Ministry of Health of The Republic of
Indonesia has received a shipment of the oral antiviral
medicine nirmatrelvir (NTV)/ritonavir (RTV)
(PAXLOVID™) (World Health Organization,
2023a). It is drug cocktail that designed to stop
COVID-19 from worsening by inhibiting the
replication of SARS-CoV-2 in patients’ body (Lin et
al., 2023). It is suitable to treat early infection in order
to stop the diseases progression. NTV is shown to be
32
Hutapea, H. M. L., Maladan, Y., Wahyono, T. Y. M., Parikesit, A. A. and Sudaryo, M. K.
The Profile of SARS-Cov-2 Genome from Indonesia and Its Impact on Paxlovid TM in Treating Covid-19.
DOI: 10.5220/0013217000003873
Paper published under CC license (CC BY-NC-ND 4.0)
In Proceedings of the 1st International Conference on Medical Science and Health (ICOMESH 2023), pages 32-37
ISBN: 978-989-758-740-5
Proceedings Copyright © 2025 by SCITEPRESS – Science and Technology Publications, Lda.

suppressing SARS-CoV-2 by binding and
suppressing Main protease (Mpro) or known as
3CLpro as GC373 analog both in vitro and in vivo.
However, it is metabolized mainly by cytochrome
P450 3A4 (CYP3A4) which is responsible for the
primary metabolism of about 50% of drugs. RTV
boosts nirmatrelvir activity by inhibiting CYP3A
(Lam & Patel, 2023).
In Coronaviridae genus, 3CLpro promotes the
replication by cleaving polyprotein when the viral
RNA enters the host cells. This protein exhibits >96%
sequence identity with SARS-CoV, and the residues
of its binding pocket are highly conserved. A study
conducted in Canada using samples collected from 1
January 2020 – 12 January 2023 shows a very low-
frequency (0.16 – 0,58%) variants at position linked
to Paxlovid
TM
resistance genes (nsp5). Even though
SARS-CoV-2 exhibit slower mutation rate (2.9×10
-
6
mutations/nt/cycle - 3.7×10
-6
mutations/nt/cycle)
(Gudiño León. et al., 2021), and 3CLpro binding
pocket residues are highly conserved, the potential of
drug resistant-associated mutation should not be
neglected.
A study was conducted to identify mutation that
naturally existed in the virus, and 100 mutations were
found in 3CLpro encoding gene. These mutations
located in NTV binding site and showed comparable
enzymatic activity to the wild-type (kcat/Km <10-
fold change) and resistance to NTV (Ki >10-fold
increase). Another spot in SARS-CoV-2 genome for
drug resistant is S144, M165, E166, H172, and Q192
which exhibit resistance 1.8 to 534.0-fold depending
on the location of the mutation (Lee et al., 2022).
Based on those data, it is important to identify the
presence of NTV resistance-related mutation to
understand the potential of drug-resistance. In this
study we aim to identify the mutation associated with
Paxlovid
TM
resistance, specifically NTV, and to
analyse the interaction between NTV with 3CLpro
mutant.
2 METHODS
2.1 Analysis of SARS-CoV-2 Genome
Profile
This was a case series study, with population was
COVID-19 patients who were eligible for SARS-
CoV-2 genome isolation according to Ministry of
Health genomic surveillance policy. We downloaded
full genome sequences submitted between Sept 1,
2022 – Oct 31, 2023 from the GISAID EpiCoV
database (https://www.gisaid.org/). We excluded the
genome if it was incomplete, and contains >5%
unidentified nucleotide. We extracted nsp5 genes
from the genomes and aligned the sequences using
MEGA X software and used SARS-CoV-2 Wuhan-
Hu isolate (NC_045512.2) as control. The data such
as patients’ gender and age, and virus’ data such as
clade lineage, and mutations were recorded from
meta data of the genome.
2.2 Molecular Docking of NTV-3CLpro
PyRx 0.9.7 integrated Autodock Vina was utilized to
perform molecular docking between NTV and
3CLPro. We were using a computer with the
following configuration ASUS ROG GL553 VE,
8GB RAM. NTV-3CLPro complex was obtained
from PDB (PDB:7SI9) and separated using saved in
PDB format. The mutant form of 3CLPro was
generated using FOLDX. Docking validation was
performed by redocking NTV to 3CLPro, and the
docking procedure was considered as valid if RMSD
value was < 2.0 Å.
3 RESULTS AND DISCUSSION
3.1 Results
3.1.1 Molecular Epidemiology of
SARS-CoV-2
There were 15,753 genomes submitted between
September 1, 2022 – October 31, 2023 and all of them
were identified as Omicron variant of SARS-CoV-2.
Among them, 14,835 genomes were having complete
genes composition, and 13,345 genomes were having
<5% of unidentified nucleotides. These genomes
were extracted from nasopharynx swab of patients
who were receiving treatment in hospital and from
genomic surveillance majority in Java Island.
Majority of them were female with age 19-45 years
old (median of age was 39 years old) (Table 1).
Table 1: Demographic and SARS-CoV-2 characteristics,
and samples distribution
Characteristics Frequency
(n=13,345)
Ag
e, Median
(
median, SD
)
39±21
Ag
e
g
rou
p
, n
(
%
)
0-18 1,553
(
11.6
)
19-45 5,631 (42.2)
46-65 3,127 (23.4)
>65 1,799 (13,5)
Missin
g
1,255
(
9.3
)
The Profile of SARS-Cov-2 Genome from Indonesia and Its Impact on Paxlovid TM in Treating Covid-19
33
Gender, n
(
%
)
Female 6,494 (48.7)
Male 5,654 (42.4)
Missin
g
1,197
(
8.9
)
Clade, n
(
%
)
GH 1
(
0.0
)
G
K
2 (0,0)
GR 7 (0,1)
GRA 13,334 (99.9)
O 1
(
0,0
)
L
inea
g
es, n
(
%
)
B.1 1,760
(
13.2
)
BA.1 651 (4.9)
BA.5 4,075 (30.5)
EG 1,117
(
8.4
)
FL 787
(
5,9
)
XBB 4,159
(
31.2
)
Others 796 (6.0)
Patients’ status, n (%)
Survive 2,717 (20.4)
Decease
d
73
(
0,5
)
Unknown 10,555
(
79.1
)
Sam
p
les’ distribution, n
(
%
)
Sumatera 1,861 (13.9)
Java 9,908 (74.3)
Bali 624
(
4.7
)
Kalimantan 604
(
4.5
)
Sulawesi 207
(
1.6
)
Nusa Tenggara 67 (0.5)
Maluku 27 (0.2)
Papua 47 (0.3)
Clade GRA was predominating which caused almost
100% the infection. In overall, subvariant XBB was
prevalent in Java, Bali, Nusa Tenggara, and Papua
while Sumatera, Kalimantan, Sulawesi, and Maluku
were dominated by BA.5 (table 2), however the
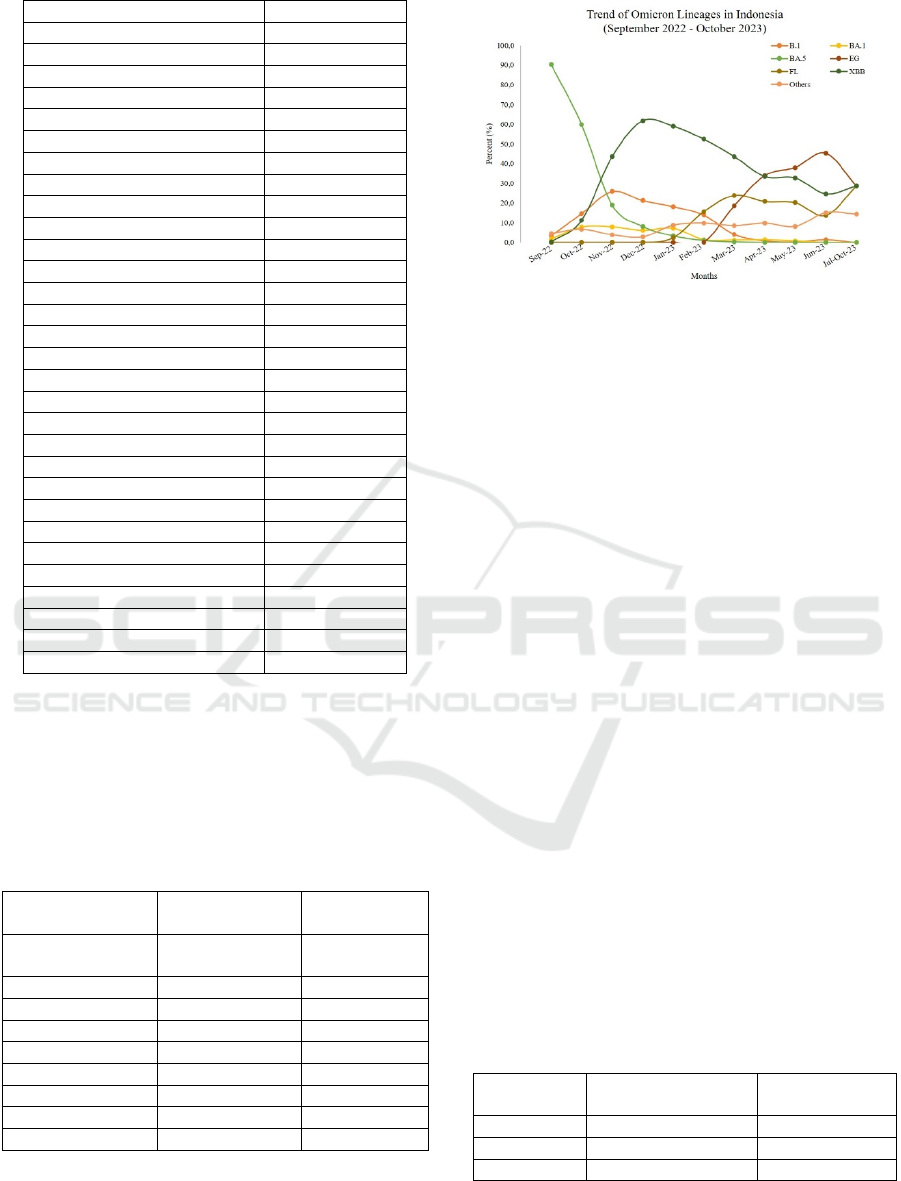
fluctuation was observed across subvariants (figure 1).
Table 2: Samples’ distribution by lineage dominance
Characteristics Frequency
(
n=13,345
)
Dominant
linea
g
e (%)
Samples’
distribution, n(%)
Sumatera 1,861
(
13.9
)
BA.5
(
47.0
)
Java 9,908
(
74.3
)
XBB
(
33.2
)
Bali 624 (4.7) XBB (39.7)
Kalimantan 604 (4.5) BA.5 (31.0)
Sulawesi 207 (1.6) BA.5 (57.0)
Nusa Ten
gg
ara 67
(
0.5
)
XBB
(
27.0
)
Maluku 27
(
0.2
)
BA.5
(
81.4
)
Pa
p
ua 47
(
0.3
)
XBB
(
42.6
)
On September 2022, the BA.5 subvariant was
predominant and causing 90.4% of the SARS-CoV-2
infection.
Figure 1: The trend of Omicron lineages in Indonesia
during the study. BA.5 lineage was dominating the
infection in the beginning of the observation, and then
replaced by EG lineage in the end of the observation.
The BA.5 infection was decreased constantly until
undetectable on January 2023. While BA.5 infection
was decreasing, XBB subvariant was increasing and
became dominant and reach its peak on December
2022. The XBB infection remained dominant for at
least a month and slowly decreased while EG
subvariant was increasing. On April 2023, the
proportion of XBB and EG subvariant was the same.
The XBB was undetected since July 2023, however
subvariant EG was taking over the infection until
October 2023.
3.1.2 Mutation Identification and Molecular
Docking
We found that 3CLpro encoding genes (nsp5) were
relatively conserved with only P132H mutation
motive (99.8%) identified. We performed molecular
docking on 3CLpro mutant P132H to NTV. In order
to obtain better understanding on the potential of
mutation to NTV susceptibility, we used 3CLpro
mutant P184H which was common in Delta variant of
SARS-CoV-2. The binding affinity of NTV-3CLpro
wild type was -8.6 kcal/mol, and the mutant P132H
and P184L were slightly weaker compared to wild
type (-8.5 kcal/mol) (Table 3).
Table 3: Docking result on NTV-3CLpro wild type and
mutants
Reseptor
Binding Affinity
(kcal/mol)
Protein stability
(kcal/mol)
Wild type -8.6 59.16
P132H -8.5 59.9
P184L -8.5 60.19
ICOMESH 2023 - INTERNATIONAL CONFERENCE ON MEDICAL SCIENCE AND HEALTH
34
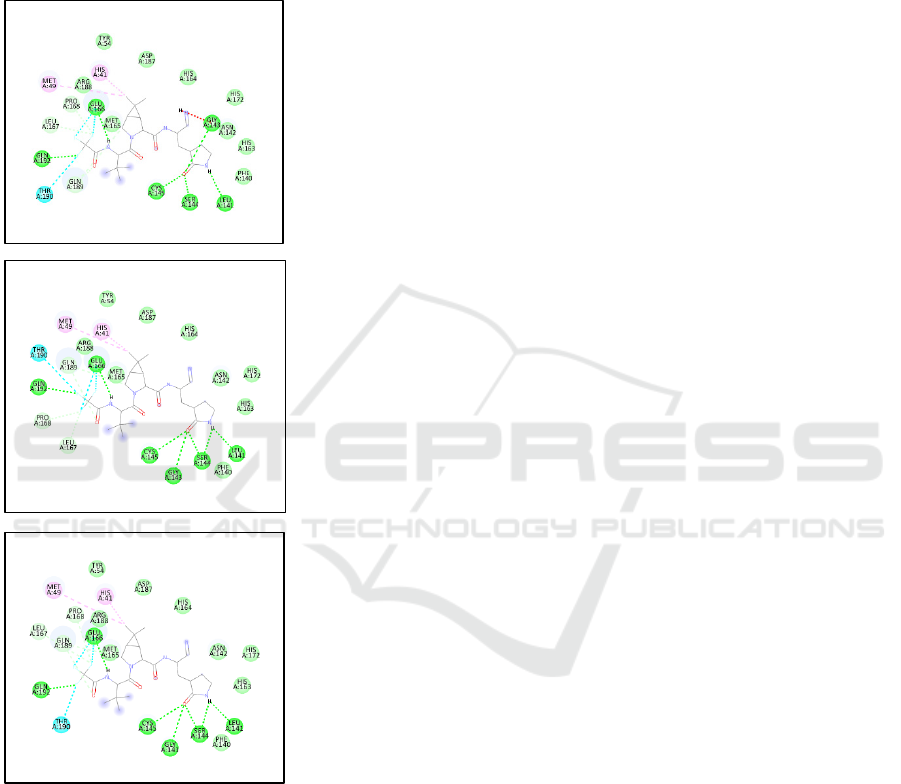
The binding affinity between 3 models was
insignificantly different, and this result was supported
by the visualization of the interaction. The hydrogen
bond was formed in the same position, which was in
amino acid LEU141, GLY143, SER144, CYS145,
GLU166, GLN192.
Figure 3: Comparison of hydrogen bond position in 3CLPro
wild type and mutant P132H in Omicron and P184L in
Delta.
3.2 Discussion
This study depicted the profile of SARS-CoV-2 and
COVID-19 patients based on metadata obtained from
GIS database. In this study, majority of the patients
(74.3%) was from Java Island, the centre of Indonesia
with highest population density (Dsikowitzky et al.,
2018). All of the patients were infected with Omicron
variants of SARS-CoV-2, majority with XBB. Study
showed that proportion of asymptomatic and mild
patients was higher in Omicron group than Delta and
Beta groups, however those with Omicron had more
throat soreness and less headache (Yang et al., 2022).
Majority of the patients was between 19-49 years old,
this was consistent with previous studies which
showed that greater proportion of Omicron infection
was found in younger age group, while Delta was
found in older age group of patients (Miao et al.,
2023; Yang et al., 2022).
COVID-19 medication was mostly involving
immunological intervention by using antibody such
as tocilizumab and other anti-SARS-CoV-2
monoclonal antibody products, however, high
variation in spike of the virus is major obstacle to
keep the medication works as the way it is (Treatment
Guidelines Panel, 2023). The discovery of 3CLpro
inhibitors represents a major breakthrough in
COVID-19 treatment (Zhu et al., 2020). In this study,
all the samples collected were identified as Omicron
variant of SARS-CoV-2, with 353 sub-lineages, and
35% of them were XBB, and followed by BA.5
(34%). Omicron variant is more transmissible and can
evade immune system better compared to previous
variants of SARS-CoV-2 (Veltri et al., 2023). The
Omicron variant have dominated epidemiologic
landscape of SARS-CoV-2 infection globally. It also
had evolved remarkably in diversity by forming over
1,000 sub-lineages with the standards lineages are
BA.1, BA.2, BA.3, BA.4, and BA.5, which share
many mutations, but also significantly different
(Velavan et al., 2023; Veltri et al., 2023).
In this study, on September 2022 we observed that
BA.5 was identified in majority of the samples. BA.5
was first detected in South Africa, and then in
Belgium, France, China, Botswana, Portugal,
Germany and Australia. The most recent ancestor of
BA.5 is estimated most closely related to BA.2. There
has been reported that BA.2 and BA.5 are more
transmissible and resistant to immunity generated by
previous variants and most monoclonal antibodies.
BA.5 dominance was dropping on October 2022 and
replaced by XBB variant. XBB was likely originated
via BA.2 descendant recombination. It was first
detected in India on October 2022 and immediately
became predominant globally. WHO declared that
XBB as Omicron subvariant under monitoring on
October 28, 2022 (Lee et al., 2022).
EG started to replace XBB since April 2023 until
October 2023. Limited information about EG.2 is
available; it is one of many subvariants that has
F456L in spike which makes it harder for many
existing antibodies to recognize the viral particles. If
Wild type
3CL-Pro
mutant P132H
3CL-Pro
mutant P184L
The Profile of SARS-Cov-2 Genome from Indonesia and Its Impact on Paxlovid TM in Treating Covid-19
35

this virus also has L455F, it might have the ability of
immune-evading and tighter binding to the ACE2
protein, which is likely to enhance cell entry. The
EG.2 variant is radically different from the others,
and Q52H and F456L mutations make this variant
more similar to the original Omicron which has
greater binding affinity compare to the other XBB
descendants (Veltri et al., 2023)
Our study showed that mutation that has potential
in developing drug resistance remains rare, however
P132H was identified in almost all samples (99.8%).
This result is in line with current global situation
where this mutation is the most prevalent globally.
P132H mutation is exclusively associated with
Omicron because this mutation was identified in
>98% of Omicron (Sacco et al., 2022). On 2022,
P132H was most prevalent in UK (44%), and it was
found in 98% of Omicron subvariant. P132H
mutation is localized at about -22 Å away from the
catalytic site and not in direct contact with any of the
residues of allosteric pocket. Study showed that
P132H got 3CLpro thermal stability compromised
(Lee et al., 2022). A study demonstrated that P132H
mutation alone related to a decreased stability of the
enzyme in vitro. Another study also revealed the
crystal structures shows that the mutations does not
give rise any significant changes of the protein around
the binding pocket or the site of the mutation
(Greasley et al., 2022).
The effect of the P132H mutation on Omicron
3CLpro remains unclear. We found that the binding
affinity of P132H mutant was not significantly
different compared to the wild type. This result was
supported by visualization of hydrogen bond
position, we observed the same position of hydrogen
bond across the mutations. Our result is consistent
with previous study which showed that P132H might
not reduce enzymatic activity and inhibitor binding.
Study showed that P132H decrease thermal stability
of 3CLpro and may cause the increasing protein
flexibility which might broaden substrate profile
substrate profile or to alter ligand binding (Sacco et
al., 2022).
Another study showed that P132H will give effect
to thermal stability only if the proline at position 108
3CLpro is also replaced by other amino acid. Another
impact of P132H was observed by Chen et al.,
Omicron with P132H mutation permitted charged L-
Lys and the enzyme activity is increased toward L-
Trp and L-Tyr compared to the wild type. Catalytic
efficiency was observed on the double mutant K90
and P132H.(Chen et al., 2023) In this study we
observed that P132H alone without other mutation
along with it, therefore P132H alone is considered to
be non-major changes related to the chemical
characteristic of 3CLpro because this mutation does
not play role whether in the active site or the allosteric
binding site of the protein (Ullrich et al., 2022).
4 CONCLUSIONS
NTV is still potent to be used as oral antiviral to treat
COVID-19 in Indonesia, however routine genomic
surveillance is necessary to anticipate the appearance
of mutations that have been proven to be associated
with NTV resistances.
ACKNOWLEDGEMENTS
We gratefully acknowledge all SARS-CoV-2
sequence data contributors in Indonesia, i.e., the
authors and their originating laboratories responsible
for obtaining the specimens, and their submitting
laboratories for generating the genetic sequence and
metadata and sharing via GISAID, on which parts of
this research is based.
REFERENCES
Chen, S. A., Arutyunova, E., Lu, J., Khan, M. B., Rut, W.,
Zmudzinski, M., Shahbaz, S., Iyyathurai, J., Moussa, E.
W., Turner, Z., Bai, B., Lamer, T., Nieman, J. A.,
Vederas, J. C., Julien, O., Drag, M., Elahi, S., Young,
H. S., & Lemieux, M. J. (2023). SARS-CoV-2 Mpro
Protease Variants of Concern Display Altered Viral
Substrate and Cell Host Target Galectin-8 Processing
but Retain Sensitivity toward Antivirals. ACS Central
Science, 9(4), 696–708.
https://doi.org/10.1021/acscentsci.3c00054
Dsikowitzky, L., Damar, A., Ferse, S. C. A., Irianto, H. E.,
Jennerjahn, T. C., Lukas, M. C., Nordhaus, I.,
Pohlmann, T., Schwarzbauer, J., Sugama, K., &
Sumiono, B. (2018). Java Island, Indonesia. In World
Seas: An Environmental Evaluation Volume II: The
Indian Ocean to the Pacific (Second Edi, Vol. 2015, pp.
459–490). Elsevier Ltd. https://doi.org/10.1016/B978-
0-08-100853-9.00029-4
Greasley, S. E., Noell, S., Plotnikova, O., Ferre, R. A., Liu,
W., Bolanos, B., Fennell, K., Nicki, J., Craig, T., Zhu,
Y., Stewart, A. E., & Steppan, C. M. (2022). Structural
basis for the in vitro efficacy of nirmatrelvir against
SARS-CoV-2 variants. Journal of Biological
Chemistry, 298(6), 1–7.
https://doi.org/10.1016/j.jbc.2022.101972
Gudiño León., A. R., Acuña López., R. J., & Terán Torres.,
V. G. (2021). Mutation rate of SARS-CoV-2 and
ICOMESH 2023 - INTERNATIONAL CONFERENCE ON MEDICAL SCIENCE AND HEALTH
36

emergence of mutators during experimental evolution.
BioRvix Pre-Print, 6.
Lam, C., & Patel, P. (2023). Nirmatrelvir-Ritonavir.
Lee, J. T., Yang, Q., Gribenko, A., Perrin, B. S., Zhu, Y.,
Cardin, R., Liberator, P. A., Anderson, A. S., & Hao, L.
(2022). Genetic Surveillance of SARS-CoV-2 Mpro
Reveals High Sequence and Structural Conservation
Prior to the Introduction of Protease Inhibitor Paxlovid.
MBio, 13(4), 1–15.
https://doi.org/10.1128/mbio.00869-22
Lin, D. Y., Abi Fadel, F., Huang, S., Milinovich, A. T.,
Sacha, G. L., Bartley, P., Duggal, A., & Wang, X.
(2023). Nirmatrelvir or Molnupiravir Use and Severe
Outcomes from Omicron Infections. JAMA Network
Open, 6(9), E2335077.
https://doi.org/10.1001/jamanetworkopen.2023.35077
Miao, Y., Ren, Y., & Ren, T. (2023). Clinical
Characteristics Profile of COVID-19 Patients with
Omicron Variant Admitted in a Tertiary Hospital,
Central China. International Journal of General
Medicine, 16(May), 2365–2371.
https://doi.org/10.2147/IJGM.S409478
Sacco, M. D., Hu, Y., Gongora, M. V., Meilleur, F., Kemp,
M. T., Zhang, X., Wang, J., & Chen, Y. (2022). The
P132H mutation in the main protease of Omicron
SARS-CoV-2 decreases thermal stability without
compromising catalysis or small-molecule drug
inhibition. Cell Research, 32(5), 498–500.
https://doi.org/10.1038/s41422-022-00640-y
Treatment Guidelines Panel. (2023). COVID-19 treatment
guidelines (p. 199).
https://www.covid19treatmentguidelines.nih.gov/
Ullrich, S., Ekanayake, K. B., Otting, G., & Nitsche, C.
(2022). Main protease mutants of SARS-CoV-2
variants remain susceptible to nirmatrelvir. Bioorganic
and Medicinal Chemistry Letters, 62(February), 13–16.
https://doi.org/10.1016/j.bmcl.2022.128629
Velavan, T. P., Ntoumi, F., Kremsner, P. G., Lee, S., &
Meyer, C. G. (2023). Emergence and geographic
dominance of Omicron subvariants XBB/XBB.1.5 and
BF.7 - the public health challenges. International
Journal of Infec, 128, 307–309.
Veltri, P., Giancotti, R., Lomoio, U., Puccio, B., Tradigo,
G., Guzzi, P., Vizza, P., & Torti, C. (2023). The
Omicron XBB.1 Variant and its Descendants: Genomic
Mutations, Rapid Dissemination and Notable
Characteristics. Authorea, 2(i).
World Health Organization. (2023a). Arrival of COVID-19
antiviral medicine helps bolster Indonesia ’ s COVID-
19 response (Issue April, pp. 12–13).
https://www.who.int/indonesia/news/detail/13-04-
2023-arrival-of-covid-19-antiviral-medicine-helps-
bolster-indonesia-s-covid-19-
response#:~:text=COVID-19 remains a global,animals
for the foreseeable future.
World Health Organization. (2023b). WHO Coronavirus
(COVID-19) Dashboard, https://covid19.who.int/
(accessed March 11, 2023).
Yang, W., Yang, S., Wang, L., Zhou, Y., Xin, Y., Li, H.,
Mu, W., Wu, Q., Xu, L., Zhao, M., Wang, C., & Yu, K.
(2022). Clinical characteristics of 310 SARS-CoV-2
Omicron variant patients and comparison with Delta
and Beta variant patients in China. Virologica Sinica,
37(5), 704–715.
https://doi.org/10.1016/j.virs.2022.07.014
Zhu, W., Xu, M., Chen, C. Z., Guo, H., Shen, M., Hu, X.,
Shinn, P., Klumpp-Thomas, C., Michael, S. G., &
Zheng, W. (2020). Identification of SARS-CoV-2 3CL
Protease Inhibitors by a Quantitative High-Throughput
Screening. ACS Pharmacology and Translational
Science, 3(5), 1008–1016.
https://doi.org/10.1021/acsptsci.0c00108
The Profile of SARS-Cov-2 Genome from Indonesia and Its Impact on Paxlovid TM in Treating Covid-19
37
