
Can HP-protein Folding Be Solved with Genetic Algorithms? Maybe not
Reitze Jansen
1 a
, Ruben Horn
2,3 b
, Okke van Eck
3 c
, Kristian Verduin
1 d
,
Sarah L. Thomson
4 e
and Daan van den Berg
1 f
1
Department of Computer Science, University of Amsterdam, Netherlands
2
Helmut-Schmidt-University, Hamburg, Germany
3
Department of Computer Science, VU Amsterdam, Netherlands
4
Napier University, Edinburgh, U.K.
Keywords:
Protein Folding, Genetic Algorithms, Evolutionary Computing, Constraints, Constraint Hierarchy.
Abstract:
Genetic algorithms might not be able to solve the HP-protein folding problem because creating random indi-
viduals for an initial population is very hard, if not impossible. The reason for this, is that the expected number
of constraint violations increases with instance size when randomly sampling individuals, as we will show in
an experiment. Thereby, the probability of randomly sampling a valid individual decreases exponentially with
instance size. This immediately prohibits resampling, and repair mechanisms might also be non-applicable.
Backtracking could generate a valid random individual, but it runs in exponential time, and is therefore also
unsuitable. No wonder that previous approaches do not report how (often) random samples are created, and
only address small instances. We contrast our findings with TSP, which is also NP-hard, but does not have
these problems.
1 PROTEIN FOLDING
The exact way in which a protein folds is immensely
important. Within the human body, the long chains
of amino acids (“aminos”) that make up our proteins
perform their biological function only when spatially
folded in a certain way. For many, their folded shape
is a state of minimum energy – of which a protein can
have multiple (Levitt, 1983; Unger and Moult, 1993).
Conversely, a folding deficiency can lead to a higher
than minimum energy, which causes the conforma-
tion to be unstable. Proteins in unstable conforma-
tions have the tendency to unfold, which is a known
cause for Alzheimer’s, Parkinson’s, diabetes, and fa-
tal insomnia (Lee et al., 2000; Thomas et al., 1995;
Dobson, 1999).
Not just for this reason the subject is intensively
studied. In medical sciences, diseases are combated
a
https://orcid.org/0009-0007-0029-2882
b
https://orcid.org/0000-0001-6643-5582
c
https://orcid.org/0000-0002-3600-5183
d
https://orcid.org/0009-0005-8754-7635
e
https://orcid.org/0000-0001-6971-7817
f
https://orcid.org/0000-0001-5060-3342
with artificially synthesized proteins, which also at-
tain their stability from their exact conformational de-
tails (Leader et al., 2008; Zhao and Lu, 2011; Don-
dapati et al., 2020). The exact mechanics of the
folding process are notoriously complex (Creighton,
1988; Dill and MacCallum, 2012), but stability result-
ing from a conformation is known to closely depend
on the interplay of the aminos (Levitt, 1982; Levitt,
1983; Dill, 1985). In exploring the nature of confor-
mations, researchers moved to utilize simplified mod-
els, such as the HP-model. Algorithmic protein fold-
ing, particularly in the HP-model, aims at finding the
maximum stability conformation of a protein, and at
understanding how to get there.
Within the HP-model, proteins are chains of con-
nected aminos which are all labeled as either being
hydrophobic (H) or polar (P) (Lau and Dill, 1989),
and precede one another on adjacent vertices in a
(usually 2- or 3-dimensional) rectangular lattice (Fig-
ure 1). Non-connected H-amino’s can form attracting
bonds when placed on neighbouring lattice vertices,
each of which reduces the free energy of the attained
conformation by one. So the more H-bonds the folded
Jansen, R., Horn, R., van Eck, O., Verduin, K., Thomson, S. and van den Berg, D.
Can HP-protein Folding Be Solved with Genetic Algorithms? Maybe not.
DOI: 10.5220/0012248500003595
In Proceedings of the 15th International Joint Conference on Computational Intelligence (IJCCI 2023), pages 131-140
ISBN: 978-989-758-674-3; ISSN: 2184-3236
Copyright © 2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)
131
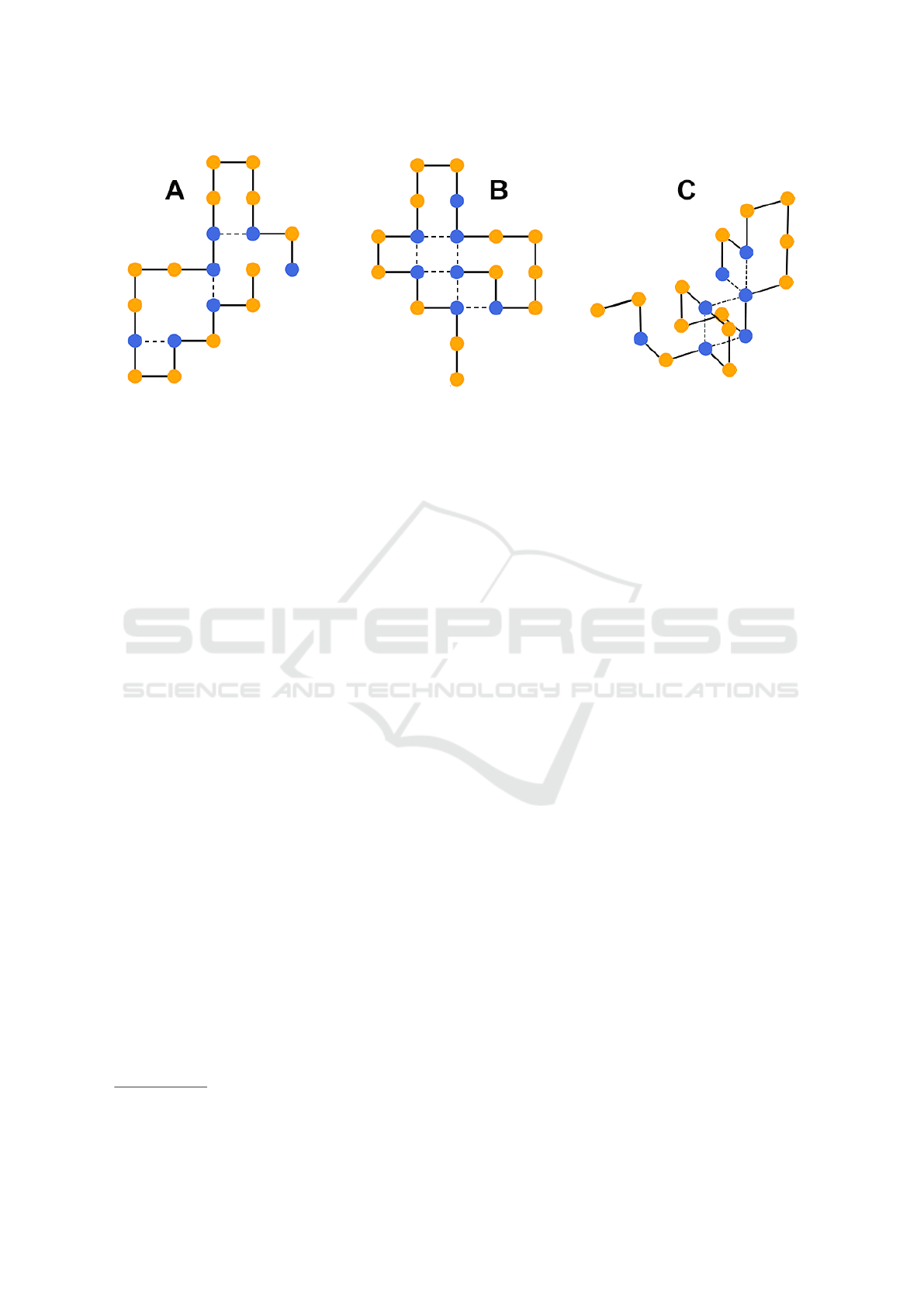
Figure 1: The HP-protein folding problem pertains finding the lowest stability conformation for a certain protein. Depending
on the fold, a protein conformation can have different stability values, which are given by the number of H-bonds (dashed
lines) of the corresponding conformation. In subfigures A and B, the protein is folded in 2D with stability -3 and -6 respec-
tively. In subfigure C, it is folded in 3D with a stability of -5.
protein has, the lower its stability value
1
and the less
likely the protein is to spontaneously unfold. The free
energy (or stability) of a conformation c for an amino
acid sequence seq = [a
1
,..,a
n
], is thereby the mini-
mization of:
S(c) =
n
∑
i=1
i−1
∑
j=1
f
n
(a
i
,a
j
)
where f
n
indicates whether a
i
and a
j
are first neigh-
boring H-aminos, defined as:
f
n
(a
i
,a
j
) =
−1
if a
i
= a
j
= H ∧ |i − j|̸= 1
∧ d(a
i
,a
j
) = 1
0 otherwise
in which d(a
i
,a
j
) is the Manhattan distance between
two aminos on the lattice (van Eck and van den Berg,
2023). Two unconnected H-aminos that are first
neighbors are said to form a bond between them, as
illustrated with the dashed lines in Figure 1. Thereby,
the HP-protein folding problem is a minimization
problem: a quest to find the conformation with the
lowest stability value, or equivalently, the highest
number of H-bonds formed between neighbouring H-
aminos.
The HP-model preserves the NP-completeness
and NP-hardness of the protein folding problem
(Berger and Leighton, 1998; Hart and Istrail, 1997),
which entails that neither finding a maximally sta-
ble conformation nor verifying a given conforma-
tion is actually maximally stable can be done in
polynomial time – under the assumption that P ̸=
1
Confusingly enough, a lower stability value means a
more stable protein
NP. Even though the HP-model is a strongly sim-
plified representation of natural protein folding, the
number of possible conformations is still immense
(Unger and Moult, 1993), and finding a minimum-
energy conformation is intractable with the capabil-
ities of modern-day computers for any realistic in-
stance size. As one consequence, researchers are fo-
cusing on (meta)heuristic approaches instead of exact
algorithms.
But this approach is not without problems ei-
ther, due to the overwhelming number of invalid in-
dividuals in the search space. The vast majority of
metaheuristic algorithms starts off with a population
of randomly chosen individuals (Eiben and Smith,
2015), and although Eiben and Smith also discuss
the possibility of nonrandom initialization, mostly for
specific problem settings, many authors will opt for a
uniformly random initial population. Maaranen et al.
take a slightly stronger position even, abiding by the
practice that initial individuals should be “as evenly
distributed as possible” (Maaranen et al., 2004). We
also closely follow this position: at the very least, ran-
dom initialization of an initial population should be
uniformly random, and unbiased. Or even more care-
fully put: at the very least we want uniformly random
sampling to be available, in order to obtain an unbi-
ased initial population for our evolutionary algorithm,
if we choose to have so.
However, even this most modest wish appears to
be a formidable problem for protein folding in the HP-
model, as we will show shortly. Uniformly randomly
sampling a conformation from the space of all pos-
sible conformations might not be possible in deter-
ministic polynomial time for this problem. To make
matters worse, stochastic uniform random sampling is
also problematic, as it appears the conformation space
ECTA 2023 - 15th International Conference on Evolutionary Computation Theory and Applications
132

gets saturated with invalid instances as n increases.
As a direct consequence, it might be impossible to
sample a uniformly random initial population, e.g. for
use by a genetic algorithm. For this reason, it is ques-
tionable how suitable genetic algorithms are for solv-
ing the HP-protein folding problem.
We will demonstrate these problems by examin-
ing 40,000 random folds for ‘neutralized’ proteins in
2D and 3D without any preassumptions, and then sim-
ply count the number of constraint violations (or col-
lisions) that occur for each protein (Figure 2). The
results show that the number of expected collisions
increases in n, making the sampling of valid random
initial individeals ever less feasible. But before that,
we will have a look at existing approaches for fold-
ing proteins with genetic algorithms. Unsurprisingly,
none of these are unproblematic.
2 EXISTING GA-APPROACHES
There have been various attempts at solving HP-
protein folding with genetic algorithms since 1993, of
which some are summarized below. Many of them are
part of the same ‘line’ of papers, initiated by Unger
and Moult in 1993 (Unger and Moult, 1993).
Generally speaking, one can represent a confor-
mation relative to the lattice (a sequence of lattice di-
rections denotes the conformation) or relative to the
chain (a sequence of left - right - up - down
turns). If the step back to the previous amino is
prevented however, we can reduce the conformation
space enormously (at least from O(4
n
) to O(3
n
)), al-
though the other representations could have imple-
mentational advantages when it comes to crossover
and mutation. In none of the cases below however,
we found any motivation for why authors opt for ei-
ther of the two representations.
The early study by Unger and Moult compared a
Monte Carlo approach to a genetic algorithm for HP-
protein folding (Kirkpatrick et al., 1983; Unger and
Moult, 1993). They use a two-dimensional Cartesian
lattice and imply that folds are encoded relative to
the chain (Patton et al., 1995). Their Monte Carlo
approach starts from a ‘random coil conformation’,
but the details of the GA’s initialization are not listed.
In the parent selection however, some details are ex-
posed: if during crossover all the three possible angles
at which to join the partial folds results in collisions,
new parents are sampled. The GA is run for 300 gen-
erations with a population size of 200, and perfectly
solves all instances with a size below 64 within less
than 600,000 evaluations. But notably, they do not re-
port the number of invalid conformations generated,
nor the number of times resampling is required in mu-
tation, crossover or initialization.
In a 1995 followup work by Patton et al., a stan-
dard GA was employed to HP-protein folding on a
3D Cartesian lattice (Patton et al., 1995). Individuals
with collisions are tolerated in the populations, but
penalized in the evaluation. Their modifications con-
sistently result in lower-energy conformations after
much fewer evaluations. So it appears that temporar-
ily allowing invalid conformations in a metaheuristic
allows for better end solutions – found quicker. But
this observation should be considered as a very early
hypothesis.
Nine years later, a followup study by Custodio et
al. implement four modifications to Patton et al.’s ap-
proach (Cust
´
odio et al., 2004). These authors conse-
quently also penalize conformations containing col-
lisions. Their selection method preserves diversity
by progressively replacing individuals with equal or
better offspring. For recombination, they use multi-
point crossover, which is contingent on the length of
the chain. They also experiment with using islands
to create a better initial population “(with fewer col-
lisions)”. Additionally, they propose an alternative
fitness function which rewards more compact con-
formations. Their representation of the folded struc-
ture is relative to the lattice. These modifications re-
sult in a better average performance and within the
3,500,000 evaluations the best solutions are almost as
good as when using the method by Unger and Moult
or Patton et al. The fact that their work is published
could be a testimony to the success of allowing col-
lisions rather than resampling, but the reader should
understand that 3.5 million evaluations is a lot.
In 2005, a study by Bui and Sundarraj on genetic
algorithms for two-dimensional HP-protein folding
appeared(Bui and Sundarraj, 2005). In this work,
secondary structures of the longest hydrophobic sub-
section were separately evolved. During random ini-
tialization, the longest hydrophobic subsection is se-
lected at random from the library of secondary struc-
tures instead of being sequentially folded. When a
collision occurs and cannot be resolved by rotation,
the individual is recreated. The top-half of the initial-
ized individuals from the first generation. Using tour-
nament selection, one-point crossover is performed to
create offspring before applying mutations across the
whole chain with a probability depending on the gen-
eration. If the mutation falls within a hydrophobic
subsection that was sampled from the library of sec-
ondary structures, it is replaced by a new secondary
structure. If possible, any collisions introduced by
crossover and mutation are resolved, or the individual
is discarded. To do this, the chain is re-folded start-
Can HP-protein Folding Be Solved with Genetic Algorithms? Maybe not
133
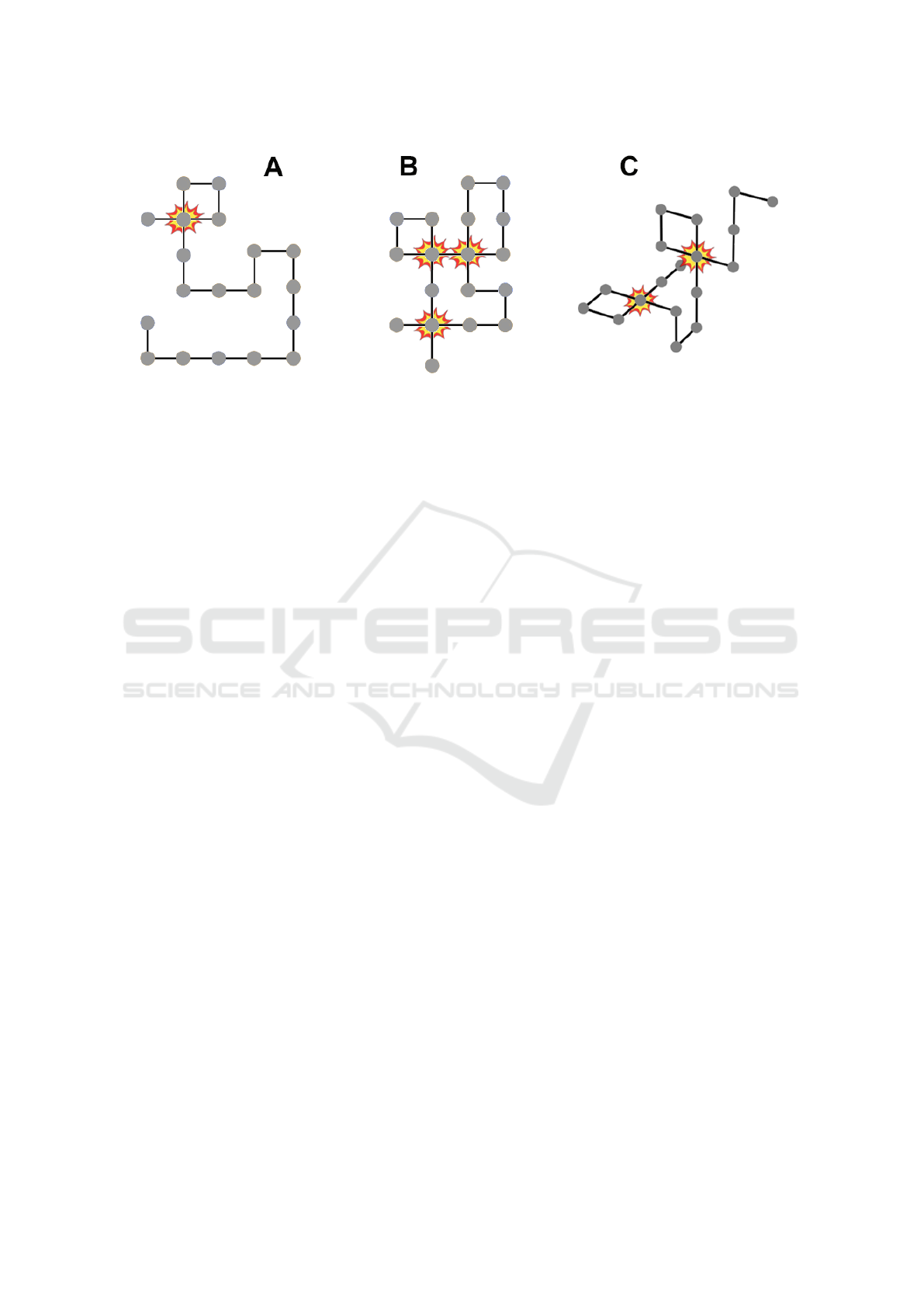
Figure 2: In HP-protein folding, finding valid conformations to evaluate is a problem. When randomly folding ‘neutral’
HP-proteins, constraint violations (or: ‘collisions’) occur quite frequently. For a protein consisting of n aminos, the minimum
number of constraint violations for any fold in 2D or 3D is 0, the maximum is n − 3.
ing from the secondary structure if present and from
the beginning if not and in the case of a collision,
a random possible direction is selected. Using two
simple refolding schemes (patterns and replacements
for 4 and 6 neighboring aminos), local optimization
is applied to the new individuals before progressively
creating the next generation by replacing the worst
parent or individual in the whole population with the
best offspring if it is better. Preceding the execution
of the main algorithm, the library of secondary struc-
tures is initialized using the same GA with some addi-
tional constraints for the longest hydrophobic subsec-
tion in the chain. Over 100,000 generations, the best
results of this approach are at least as good as those
by Unger and Moult’s and also (very close to) per-
fect for selected instances with a length of up to 100.
Summarizing, this work is another example of how
many tailor-made repair mechanisms are required to
solve HP-protein folding. These authors have one for
initialization, crossover and mutation, and by reading
their study, there’s no guarantee the algorithm won’t
get stuck on trying to find zero-collision conforma-
tions. True, these might practically not occur often
at all, but as we will show, the proportion of invalid
inviduals in the conformation space grows as n in-
creases, so this is a problem worth of recognition.
Bui and Sundarraj themselves give no numbers on of-
ten their resampling methods are invoked, and if they
ever get stuck, for either initialization, mutation, or
crossover.
Four years later, a hybrid approach by Lin and
Hsieh involved a genetic algorithm combined with
the Taguchi method and particle swarm optimization
(Lin and Hsieh, 2009) (for the Taguchi method see
(Kaytako
˘
glu and Akyalc¸ın, 2007)). Following repro-
duction using roulette-wheel selection and two-point
crossover using tournament selection, the Taguchi
method using random pairs replaces the bottom-half
of the population. A random subsection of the chain
is selected for mutation, on which a Particle Swarm
Optimization (PSO) algorithm is applied, but crucial
details such as the maximum number of its iterations
are not mentioned. Finally, hill climbing is performed
on a random subsection of the chain. Their represen-
tation of the folded structure is also relative to the lat-
tice, but the treatment of collisions is not described.
Over 2,000 generations and using a population of be-
tween 100 and 300, their algorithm consistently finds
lower-energy conformations than the experiment by
Unger and Moult, however the number of function
evaluations is not compared directly. A fair conclu-
sion for now is to say we do not know enough about
their experiment to draw direct conclusions, but we
suspect at the very least that authors also did not solve
the collision problem. Fairer would be to say that this
experiment needs to be replicated to obtain useful data
about their collision number and treatment.
A different approach, using multi-objective opti-
mization is proposed by Garza-Fabre et al. (Garza-
Fabre et al., 2015). Their experiment also allows for
individuals with collisions whose number, along with
the number stability of the conformations, make up
the two optimization criteria. Through neighboring
non-dominated solutions, it may be possible to ex-
plore the search space more effectively and escape
them. Their experiments on both two- and three-
dimensional Cartesian lattices are encoded relative
to the chain, and these authors find that (proportion-
ally) biasing the optimization criteria to favor a re-
duction in collisions is necessary and yields better re-
sults compared to single-objective optimization. The
advantage of this approach becomes clear with in-
creased evaluations. However, this approach still re-
sults in searching a very large state space containing
mostly invalid conformations, and hence an efficient
restriction of the search space to only those without
ECTA 2023 - 15th International Conference on Evolutionary Computation Theory and Applications
134

collisions would be preferable. Nonetheless, we feel
that the bias values in their experiment hold the po-
tential to elucidate the fundamentals of this problem,
and are therefore of significant interest.
A 2016 study by Wang et al. proposed a ge-
netic algorithm with cloning of top individuals and
crossover followed by ‘chaotic mutation’ for the re-
maining individuals in the population (Wang et al.,
2016). Their conformation representation is relative
to the chain. They do not describe how the random
initialization of the population or the genetic opera-
tors treat collisions, but these authors claim their ap-
proach is significantly more likely to find the “per-
fect” conformation compared to standard GAs, even
for some long chains.
In a recent study on a 2D triangular lattice, au-
thors combine hill climbing and tabu search in a hy-
brid genetic algorithm, somewhat similar to the stud-
ies by Unger & Moult and Lin & Hsieh (Boumedine
and Bouroubi, 2021). The initial population is created
randomly, and collisions are resolved by resampling
the individual but again, authors do not mention how
often this happens. Offspring are generated by ei-
ther mutation or tabu search with random single point
crossover followed by hill climbing. This makes their
GA non-standard, and the frequency and treatment of
invalid conformations is not reported. Abiding by an
elitist approach, only the best offspring is considered
for the new population, which does not contain dupli-
cates. The use of local and tabu search makes it im-
possible to gauge the number of evaluations. Again,
we feel that there is no conclusive evidence about the
sampling distribution, which might be worth a closer
look.
The last study discussed in this paper is by Atari
and Majd, who introduce a ‘quantum genetic algo-
rithm’ (Atari and Majd, 2022). Their conformation
encoding is relative to the lattice, using two qubits
for every subsequent amino acid. The first population
is initialized with equal probability for every confor-
mation. The population is sampled from the quan-
tum state by comparing a random variable to the am-
plitude of each qubit. Using the best individual, the
quantum population is updated by aligning it with the
best sampled individual. If the algorithm converges
on a local optimum, some individuals are randomly
recreated to induce diversity. This approach implies
the penalization of conformations with collisions, but
this is not described explicitly. We had some trouble
getting the details of this this study explicit, but as
the most recent publication, it is nonetheless included
in the overview. A definite answer of how (random)
sampling occurs, and how and how often collisions
were treated was not found however.
A rather disturbing observation from these studies
is that many of these limit the length n to 64. In some,
the length is set to n ≤ 100, but this is nowhere near
the length of real proteins, which can contain up to
2,000 aminos (Alberts et al., 2003). Could it be that
not only random sampling is impossible, but iterative
algorithms as a whole should be disqualified for the
HP-protein folding problem?
3 SOLUTION SAMPLING
Reviewing the literature on the subject, it appears safe
to say that sampling the conformation space presents
a stubborn problem for genetic algorithm approaches
on HP-protein folding. In our view, the issue boils
down to the following three questions:
1. Is it possible to sample, with uniform probabil-
ity, a single conformation from the space of all
possible valid conformations, for the HP-protein
folding problem?
2. If this is possible in deterministic time, what time
complexity does such a uniform probability sam-
pling algorithm have?
3. If this is not possible, what is the (stochastically
expected) number of resamples needed to obtain
a randomly sampled conformation, and how does
this number scale in n?
We think the answers to these questions might have
far reaching consequences. For now, it appears there
are two common strategies in publications up to this
point.
1. One option is to include only valid solutions in
the search space, by rejecting, preserving and re-
pairing strategies (Unger and Moult, 1993; Bui
and Sundarraj, 2005; Boumedine and Bouroubi,
2021; Garza-Fabre et al., 2015). There are at least
three problems with this approach:
(a) Repair mechanisms such as found in the
above literature, could distort the uniformity of
the sample distribution from the conformation
space. Put differently: repairing invalid sam-
ples might bias the sample, and stochastically
miss good or even optimal solutions as a result.
(b) The expected number of required samples
might increase in n for HP-protein folding. For
some problems, such as the traveling tourna-
ment problem, this number in fact increases so
fast that the random sampling method in itself
becomes unfeasible, let alone the accompany-
ing genetic algorithm (Verduin et al., 2023b).
Possibly for this reason, most above studies
Can HP-protein Folding Be Solved with Genetic Algorithms? Maybe not
135

have n ≤ 64, and only 3 studies have instances
of n ≤ 100 aminos (Bui and Sundarraj, 2005;
Wang et al., 2016; Boumedine and Bouroubi,
2021). Most proteins in nature are at least
twice as long, but can range up to 2,000 aminos
in length (Alberts et al., 2003). Considering
the exponential growth of the number of con-
formations in n, serious doubt could be raised
on whether one can uniformly sample random
real-life sized instances even in the vastly sim-
plified HP-model, let alone solve them with
a genetic algorithm. One key insight should
come from an assessment pertaining how the
ratio of valid to invalid conformations develops
in the exponential conformation space growth.
Probably not very good, as we will shortly see.
(c) In case a deterministic uniform sampling algo-
rithm exists, such as a form of backtracking, the
time complexity of the sampling procedure be-
comes an immediate concern. Maybe for that
reason, we did not find a single study applying
that approach. It is an open question whether
a deterministic polynomial time uniform sam-
pling algorithm exists.
2. Alternatively, one might allow but penalize
conformation collisions (Patton et al., 1995;
Cust
´
odio et al., 2004; Garza-Fabre et al., 2015),
which potentially allows a genetic algorithm to
converge on a better conformation via invalid
ones. However, the search space size increases
dramatically, as invalid conformations become
ever more common in n, possibly making the en-
tire experiment unfeasible. Additionally, the pe-
nalization strategy for collisions presents yet an-
other optimization problem (Runarsson and Yao,
2000). Conversely, this approach might conserve
the connectedness of the fitness landscape, possi-
bly unbiasing search algorithms, which might be
beneficial.
So far, the emerging image is not a comfortable one.
Is it
¨
uberhaupt possible to randomly sample from the
conformation space with uniform probability for the
HP-protein folding problem? Maybe not. But if so,
what are the (expected) time budgets involved for dif-
ferent protein lengths n? And how does this influ-
ence the operation of GAs on the problem, or restrict
crossover and mutation operators? So many ques-
tions.
In this paper, we will start the quest for answers
by simply counting the number of collisions for dif-
ferent values of n, and rigorously characterizing them.
Although the experimental setup is quite modest, the
results could be quite fundamental.
4 EXPERIMENT & RESULTS
In the experiment, we randomly fold 1000 ‘neutral-
ized’ proteins for lengths n ∈ {5,10,15...195,200} on
both a 2D lattice and a 3D lattice without any pre-
assumptions, and counted the number of collisions
(Fig. 2). ‘Neutralize’ means we simply stripped the
amino’s ‘hydrophobic’ and ‘polar’ labels, to empha-
size we are only interested in valid conformations,
not optimal ones (Fig. 2). We place the aminos one
by one, every subsequent placement randomly choos-
ing a location ∈ {left, right, straight} for
2D, and ∈ {left, right, straight up, down}
for 3D. As such, we abide by a chain-relative repre-
sentation, but excluding backward collisions. While
placing, we increment the number of constraint vio-
lations (n
violations
) every time an amino is placed on a
lattice vertex that already has one or more previously
placed aminos.
The minimum value for n
violations
of any confor-
mation is 0, in case the random result happens to be
a completely self-avoiding walk, while the maximum
value for n
violations
is n − 4, because it’s impossible to
place the first 4 aminos on a previously occupied lat-
tice vertex, after which every k
th
amino can be placed
on top of the (k − 4)
th
amino. As a consequence, the
maximum-collision conformation has the shape of a
layered coil, and there are exactly 2 such conforma-
tions: one consisting of left turns only, and the other
consisting of right turns only. The number of zero-
violation conformations on the other hand, is much
higher than 2. As one indication, a zero-violations
conformation with exactly one left turn followed by
zero or more straights and then exactly one right
turn is (n − 2) · (n −3), for all n > 3 (since having two
turns in a conformation of just 3 aminos is impossi-
ble). This observation is important, because it imme-
diately gives rise to the suspicion of an asymmetric
distribution for the number of violations. This suspi-
cion is empirically corroborated in our results, which
will be discussed shortly. All generation was done on
a contemporary desktop computer, and took at most
three days of continuous running time. Our Python
source code is publicly available (Anonymous, 2023).
For every length n, the number violations for each
of the 1,000 conformations was recorded, and taken
to histogram (Figure 3). It turns out that these vio-
lation distributions can be closely characterized by a
beta-binomial function, which can be used to model
probability densities for discrete but finite numbers of
interdependent events. To which extent the distribu-
tion is a theoretically credible model for random con-
formations in the HP-model is debatable. For safety,
let’s just say we use this specific curve because it gen-
ECTA 2023 - 15th International Conference on Evolutionary Computation Theory and Applications
136
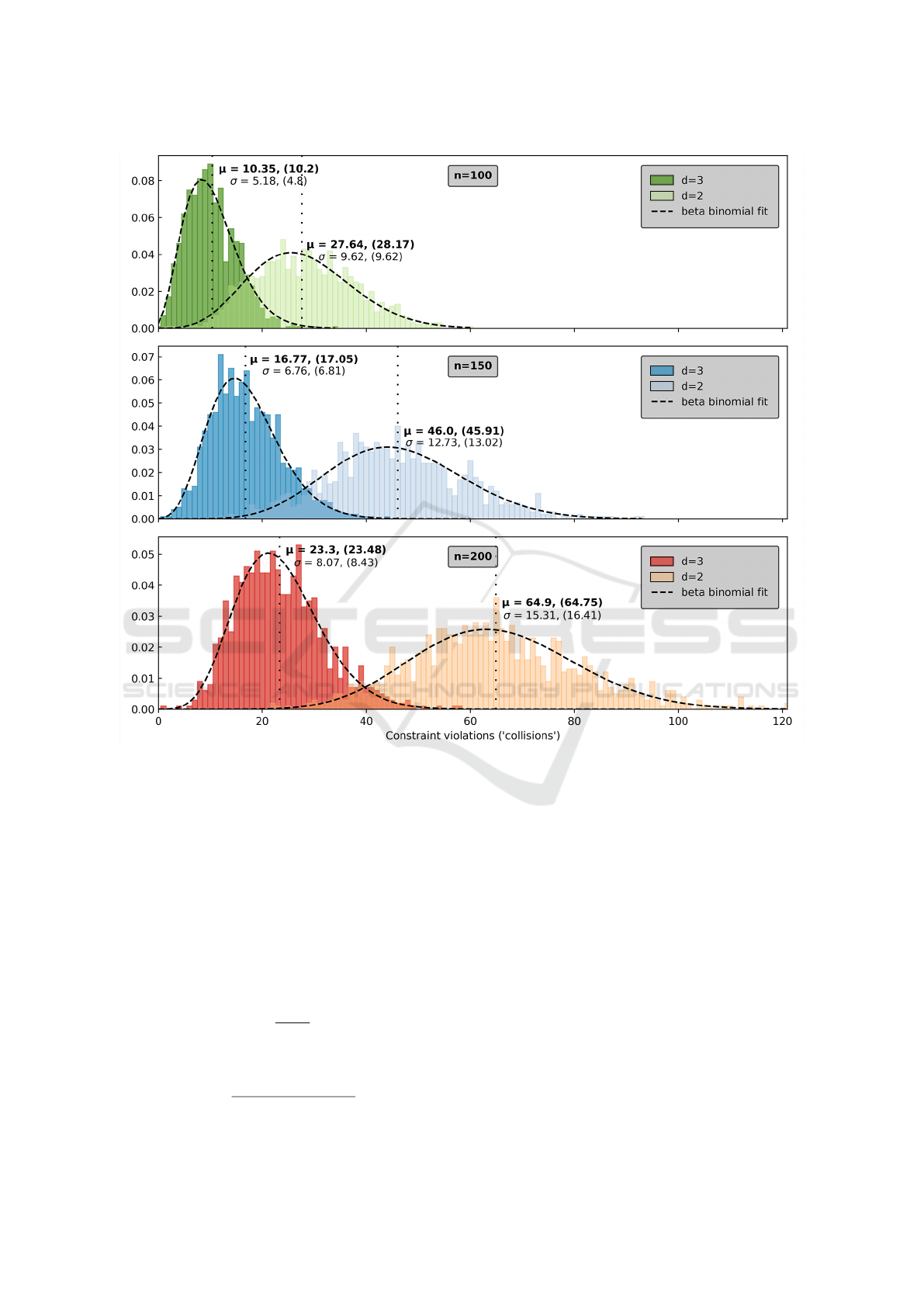
Figure 3: The frequency of constraint violations (‘collisions’) for random protein conformations of length n = 100, 150,200
in both 2 and 3 dimensions increase with n. Vertically dotted lines denote the mean (µ) of the fitted distribution, the value in
brackets is the actual value of µ taken from the data, and similar for σ.
eralizes so well, and thereby allows us to extrapolate.
The beta binomial function takes two fit-
parameters α and β, which have no predictive signifi-
cance by themselves, but rather control the skewness.
Fits were made using Python’s SciPi package and
were relatively tight (an exhaustive list of all param-
eters can be found in the repo: (Anonymous, 2023)),
but typically, larger values of n have tighter fits. Once
the fits have been made, a generalized µ and σ can be
extracted as
µ =
nα
α + β
(1)
and
σ =
nαβ(α + β + n)
(α + β)
2
(α + β + 1)
(2)
Subsequently, µ and σ themselves can also be char-
acterized in n, which can be seen in Figure 4. The
first two subfigures reveal that the number of expected
constraint violations increases linearly in the length of
the proteins (Figure 4, left) (fit details for all param-
eters can be found in the repo: (Anonymous, 2023)).
For 2D random folds, the expected number of con-
straint violations behaves like
n
violations
= 0.3678n (3)
whereas in 3D random folds, the number of constrain
violations behaves like:
n
violations
= 0.1295n (4)
Although a linear increase is quite innocuous in many
problem solving contexts, in this case it is quite se-
vere. These numbers show an increase in expected
Can HP-protein Folding Be Solved with Genetic Algorithms? Maybe not
137
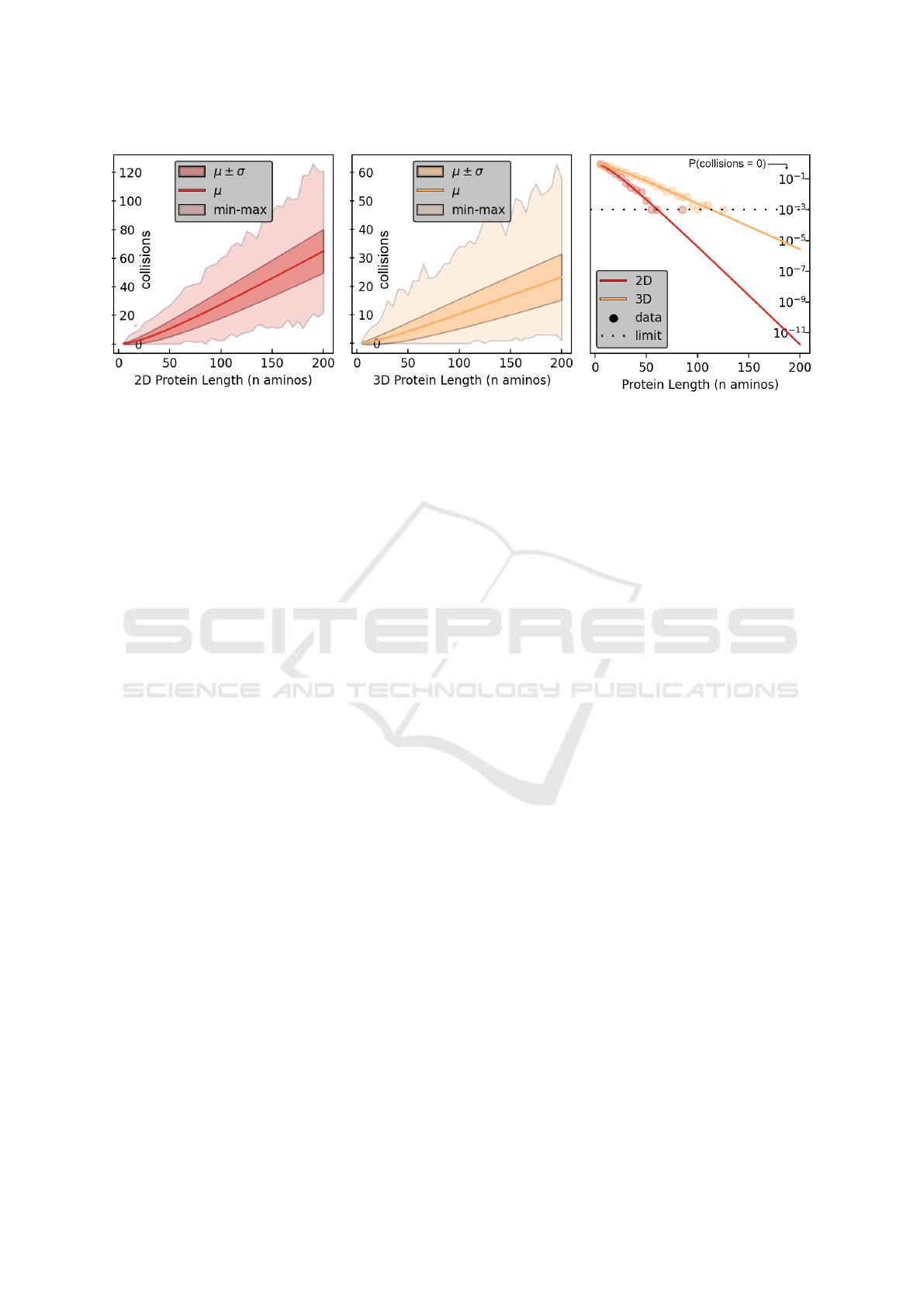
Figure 4: Left, Middle: The number of expected constraint violations (collisions) in folded protein conformations increases
linearly in the protein length, but slower in 3D than in 2D. Right: The chance of randomly sampling a zero-collision confor-
mation drops exponentially in n, but slightly slower in 3D than in 2D.
collisions as proteins get longer, and that might ex-
plain why none of the previous studies actually ran-
domly sample from the conformation space (without
problems): it simply cannot be done. A very, very pe-
culiar observation is that the 0.3678 coefficient of the
2D violations is very close to 1/e.
In the classic HP-protein folding problem, for any
random conformation to be evaluated, it needs to have
zero violations, and this probability rapidly drops to
zero for larger values of n (Figure 4, right subfigure).
The probability of sampling a zero-violation confor-
mation in 2D is
P
(violations=0)
= 2.6024 · e
−0.1294n
(5)
which is a disheartening decrease in n. For 3D, the
probability of sampling a zero-violation conformation
is
P
(violations=0)
= 1.7629 · e
−0.0654n
(6)
which is less bad, but still not good. Presumably, the
dimensional increase, and thereby the degrees of free-
dom press both constants a bit, increasing the chance
of zero violations for random folds in 3D, as com-
pared to 2D.
5 CONCLUSION & DISCUSSION
Although the setup of this experiment is quite mod-
est, the implications could be huge. With an expected
number of collisions increasing in n, and a zero-
collision sample rapidly decreasing in n, it is very
hard, if not impossible, to sample valid initial solu-
tions for lengths up to n = 200, let alone more biolog-
ically realistic lengths of up to n = 2000. This result
reveals a rather disturbing reality: iterative heuristic
algorithms, a rather weak class in the spectrum, might
not be usable for HP-protein folding with any realistic
value of n, simply because we cannot uniformly ran-
domly sample from the conformation space. More-
over, we also suspect that the trouble with crossover
and mutation operators stem from the same source:
too many constraint violations. For smaller numbers
of n, some (possibly non-uniform) sample-and-repair
methods exist, which might or might not compro-
mise the metaheuristic’s performance, but for larger
numbers of n, there might not exist any reasonable
method of randomly generating initial solutions. This
just adds up to the fact that HP-protein folding is al-
ready NP-hard, thereby also disqualifying the elite
class of exact algorithms, even for lengths of ‘only’
200 aminos.
These conclusions paint a bleak picture for the
problem’s future, but there might be some ways for-
ward. Ultimately, we want to know whether it is
possible to uniformly randomly sample a conforma-
tion for the HP-protein folding problem in determin-
istic time, and what the lowest possible time com-
plexity for that random sampling algorithm would be.
Maybe, despite previously mentioned efforts, such a
sampling algorithm exists, but has simply never been
found yet. The same might be true for universally
applicable crossover and mutation operators; maybe
these still exist, but remain to be designed. Further-
more, problems like n-queens and traveling salesman
have proven to heavily rely on the existence of an effi-
cient representation; considering the diversity in ear-
lier studies on HP-protein folding, this might be an-
other line worth exploring.
So things look quite disheartening, but things
could be worse. As it turns out, the traveling tourna-
ment problem also does not have a uniform sampling
method (yet), other than recreating invalid individu-
ECTA 2023 - 15th International Conference on Evolutionary Computation Theory and Applications
138

als. And for this problem, the constraint violations
increase quadratically instead of linearly, becoming
infeasible from n = 12 already (Verduin et al., 2023b;
Verduin et al., 2023a). But things could be better, too.
For a problem such as TSP, uniformly randomly sam-
pling can be done in deterministic linear time (θ(n)):
1. Start with a full list of unpicked cities, and an
empty tour.
2. Add a randomly picked city from the list of un-
picked cities, and add it to the tour.
3. Delete that city from the list of unpicked cities.
4. If the list of unpicked cities is not empty, go back
to 2.
While this algorithm might be considered too triv-
ial to explicitly write down, it important to realize
that this method produces a uniformly random valid
TSP-solution in deterministic linear time. Point 3 is
important in this sense. Many programmers opt for
a boolean ‘picked marker’ for each city, and sim-
ply pick a new random city when an already chosen
city is accidentally picked. This will work for up to
very large instances without any noticeable delays,
and might even stochastically improve runtimes, as
deletion from a data structure such as an array is ex-
tremely expensive compared to flipping a bit in a list
of boolean picked markers.
So it exists for TSP, but does such a uniform prob-
ability linear time selection algorithm also exist for
NP-protein folding? We do not think so. The best
we can do (for now) appears to be randomly sampling
a conformation by assigning all aminos a random di-
rection ∈ {left, right, straight}, relative to the
chain. Although this does guarantee that all confor-
mations have equal probability, it also includes a lot
of invalid conformations with colliding aminos. The
obvious solution is just to resample a few times until
we pick a valid solution, but how feasible is this ap-
proach as instances get bigger? Not very feasible, it
seems. For now, the race is on to find a determinis-
tic polynomial time uniform sampling method, which
might or might not exist. For the future of this prob-
lem, it is of utmost importance.
REFERENCES
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K.,
Walter, P., and Chaffey, N. (2003). Molecular biology
of the cell. 4th ed. Oxford University Press.
Anonymous (2023). Repository containing source
material: https://anonymous.4open.science/r/PF-
sampling/README.md.
Atari, M. and Majd, N. (2022). 2d hp protein folding us-
ing quantum genetic algorithm. In 2022 27th Inter-
national Computer Conference, Computer Society of
Iran (CSICC), pages 1–8.
Berger, B. and Leighton, T. (1998). Protein folding in the
hydrophobic-hydrophilic (hp) is np-complete. In Pro-
ceedings of the second annual international confer-
ence on Computational molecular biology, pages 30–
39.
Boumedine, N. and Bouroubi, S. (2021). A new hybrid
genetic algorithm for protein structure prediction on
the 2d triangular lattice. Turkish Journal of Electrical
Engineering and Computer Sciences, 29(2):499–513.
Bui, T. N. and Sundarraj, G. (2005). An efficient genetic al-
gorithm for predicting protein tertiary structures in the
2d hp model. In Proceedings of the 7th Annual Con-
ference on Genetic and Evolutionary Computation,
GECCO ’05, page 385–392, New York, NY, USA. As-
sociation for Computing Machinery.
Creighton, T. E. (1988). The protein folding problem. Sci-
ence, 240(4850):267–267.
Cust
´
odio, F. L., Barbosa, H. J. C., and Dardenne, L. E.
(2004). Investigation of the three-dimensional lattice
hp protein folding model using a genetic algorithm.
Genetics and Molecular Biology, 27(4):611–615.
Dill, K. A. (1985). Theory for the folding and stability of
globular proteins. Biochemistry, 24(6):1501–1509.
Dill, K. A. and MacCallum, J. L. (2012). The
protein-folding problem, 50 years on. science,
338(6110):1042–1046.
Dobson, C. M. (1999). Protein misfolding, evolution and
disease. Trends in biochemical sciences, 24(9):329–
332.
Dondapati, S. K., Stech, M., Zemella, A., and Kubick,
S. (2020). Cell-free protein synthesis: a promis-
ing option for future drug development. BioDrugs,
34(3):327–348.
Eiben, A. E. and Smith, J. E. (2015). Introduction to evolu-
tionary computing. Springer.
Garza-Fabre, M., Rodriguez-Tello, E., and Toscano-Pulido,
G. (2015). Constraint-handling through multi-
objective optimization: The hydrophobic-polar model
for protein structure prediction. Computers & Opera-
tions Research, 53:128–153.
Hart, W. E. and Istrail, S. (1997). Robust proofs of np-
hardness for protein folding: general lattices and en-
ergy potentials. Journal of Computational Biology,
4(1):1–22.
Kaytako
˘
glu, S. and Akyalc¸ın, L. (2007). Optimization of
parametric performance of a pemfc. International
Journal of Hydrogen Energy, 32(17):4418–4423.
Kirkpatrick, S., Gelatt, C. D., and Vecchi, M. P. (1983).
Optimization by simulated annealing. Science,
220(4598):671–680.
Lau, K. F. and Dill, K. A. (1989). A lattice statistical me-
chanics model of the conformational and sequence
spaces of proteins. Macromolecules, 22(10):3986–
3997.
Can HP-protein Folding Be Solved with Genetic Algorithms? Maybe not
139

Leader, B., Baca, Q. J., and Golan, D. E. (2008). Protein
therapeutics: a summary and pharmacological classi-
fication. Nature reviews Drug discovery, 7(1):21–39.
Lee, C., Park, S.-H., Lee, M.-Y., and Yu, M.-H. (2000).
Regulation of protein function by native metastabil-
ity. Proceedings of the National Academy of Sciences,
97(14):7727–7731.
Levitt, M. (1982). Protein conformation, dynamics, and
folding by computer simulation. Annual review of bio-
physics and bioengineering, 11(1):251–271.
Levitt, M. (1983). Protein folding by restrained energy min-
imization and molecular dynamics. Journal of molec-
ular biology, 170(3):723–764.
Lin, C.-J. and Hsieh, M.-H. (2009). An efficient
hybrid taguchi-genetic algorithm for protein fold-
ing simulation. Expert Systems with Applications,
36(10):12446–12453.
Maaranen, H., Miettinen, K., and M
¨
akel
¨
a, M. M. (2004).
Quasi-random initial population for genetic algo-
rithms. Computers & Mathematics with Applications,
47(12):1885–1895.
Patton, A. L., Punch, W. F., and Goodman, E. D. (1995). A
standard ga approach to native protein conformation
prediction. In Proceedings of the 6th International
Conference on Genetic Algorithms, page 574–581,
San Francisco, CA, USA. Morgan Kaufmann Publish-
ers Inc.
Runarsson, T. and Yao, X. (2000). Stochastic ranking for
constrained evolutionary optimization. IEEE Trans-
actions on Evolutionary Computation, 4(3):284–294.
Thomas, P. J., Qu, B.-H., and Pedersen, P. L. (1995). De-
fective protein folding as a basis of human disease.
Trends in biochemical sciences, 20(11):456–459.
Unger, R. and Moult, J. (1993). Genetic algorithms for pro-
tein folding simulations. Journal of Molecular Biol-
ogy, 231(1):75–81.
van Eck, O. and van den Berg, D. (2023). Quantifying
instance hardness of protein folding within the hp-
model. (accepted for publication at CIBCB’23).
Verduin, K., Thomson, S. L., and van den Berg, D.
(2023a). Too constrained for genetic algorithms. too
hard for evolutionary computing. the traveling tourna-
ment problem (to appear at ecta 2023).
Verduin, K., Weise, T., and van den Berg, D. (2023b). Why
is the traveling tournament problem not solved with
genetic algorithms?
Wang, S., Wu, L., Huo, Y., Wu, X., Wang, H., and Zhang,
Y. (2016). Predict two-dimensional protein folding
based on hydrophobic-polar lattice model and chaotic
clonal genetic algorithm. In Yin, H., Gao, Y., Li, B.,
Zhang, D., Yang, M., Li, Y., Klawonn, F., and Tall
´
on-
Ballesteros, A. J., editors, Intelligent Data Engineer-
ing and Automated Learning – IDEAL 2016, pages
10–17, Cham. Springer International Publishing.
Zhao, L. and Lu, W. (2011). D-peptide-based drug discov-
ery aided by chemical protein synthesis. Israel Jour-
nal of Chemistry, 51(8-9):868–875.
ECTA 2023 - 15th International Conference on Evolutionary Computation Theory and Applications
140
