
Data Analytics Framework for Identifying Relevant Adverse Events in
Medical Software
Md Moin Uddin and Mouzhi Ge
European Campus Rottal-Inn, Deggendorf Institute of Technology, Deggendorf, Germany
Keywords:
Clinical Evaluation, Adverse Event, Medical Software, Natural Language Processing, Machine Learning.
Abstract:
The clinical evaluation process is an ongoing and iterative process. Through clinical evaluation, the clinical
performance and effectiveness of the medical device will be monitored. While the clinical evaluation process
requires clinical data, these relevant data may come from different sources. One of the recommended sources
is “medical device adverse event report database”, which is mentioned in several guidance documents, since
the adverse event reports are useful to identify hazards caused by substances or technologies used in medical
devices. They also contain signals on the new or unknown risks associated with medical devices. As the use
of medical devices is increasing, new adverse event reports are being updated in a daily manner, thus, the size
of the adverse event database is also increasing. It is difficult and time-consuming to collect and process data
from multiple adverse event data sources and feed the data into the clinical evaluation process needs special
consideration. In this paper, the feasibility of adopting a data analytic ecosystem to deal with a large amount
of adverse event data is studied. As an output, a data analytics framework is proposed to process and classify
adverse event texts. The whole process will significantly facilitate the clinical evaluation process for medical
image analysis software.
1 INTRODUCTION
The healthcare industry is being transformed by tech-
nological advancement. The use of software in the
health sector is significantly increasing over the last
decade. On the one hand, it is making health ser-
vices efficient and on the other hand, it is, however,
creating various types of risks. The use of Artifi-
cial Intelligence (AI) in the medical decision-making
process has created new challenges for us. For ex-
ample, the “Threac -25” incident (Leveson, 1995) is
caused by software errors that pose risks to patients.
In addition, heterogeneous medical device regulations
and approval procedures across the world also cre-
ate various challenges for medical device manufac-
turers. Since the application of better data analytics
and making data-driven decisions may solve some of
these challenges (Chen et al., 2017), this paper be ex-
ploring how medical device adverse event data can be
used to make medical software safe and effective.
The complexity of medical device regulations has
been increasing over time (Agyei et al., 2022). The
goal is to make medical devices safe and effec-
tive. Also, the application of AI technology in med-
ical software makes the product even more complex.
Medical device regulation plays an important role in
the maintenance of the safety and performance of the
medical device. As part of medical device regulatory
requirements, manufacturers have to conduct differ-
ent tasks such as clinical evaluation, and post-market
surveillance activity. Most of these activities take
place in both the pre-and post-marketing stage of a
medical product’s life cycle and often rely on clinical
data (Nadakinamani et al., 2022).
The term “Clinical Data” has been defined and
mentioned in different medical device regulatory
guidance documents(Astapenko, 2019; Commission,
2007) and refers to “Safety, clinical performance
and/or effectiveness information that is generated
from the clinical use of the medical device.” Clini-
cal data may come from multiple sources (Astapenko,
2019; Kessler, 2007; GHTF, 2012) and reside in dif-
ferent forms and locations. This data may include
relevant published literature, post-market surveillance
data, adverse event databases, and recall databases.
As per regulatory guidance documents, the manufac-
turer is the responsible entity to identify relevant clin-
ical data. However, it is a very time-consuming pro-
cess to collect clinical data as the source of clinical
data is diverse. In fact, most of the clinical data lives
Uddin, M. and Ge, M.
Data Analytics Framework for Identifying Relevant Adverse Events in Medical Software.
DOI: 10.5220/0012038900003476
In Proceedings of the 9th International Conference on Information and Communication Technologies for Ageing Well and e-Health (ICT4AWE 2023), pages 81-90
ISBN: 978-989-758-645-3; ISSN: 2184-4984
Copyright
c
2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)
81

in text format. A considerable amount of effort is re-
quired to appraise and utilize this data for a medical
device regulatory task such as clinical evaluation.
This paper, therefore, aims to build a text clas-
sifier that would classify clinical data based on rel-
evancy to the medical software in question. It will
be focused on medical image analysis software and
the goal is to identify relevant adverse event data to
support the clinical evaluation process for this soft-
ware. According to European Medical Device Regu-
lation (MDR), “Clinical Evaluation” is, a systematic
and planned process to continuously generate, col-
lect analysis and assess the clinical data pertaining
to a device in order to verify the safety and perfor-
mance including clinical benefits, of the device when
used as intended by the manufacturer (European Par-
liament and Council of the European Union, 2017).
The key objectives of clinical evaluation include peri-
odically monitoring safety, and performance, also as-
sessing the risk-benefit of medical devices. Figure 1
illustrates a conceptual workflow to conduct a clinical
evaluation for medical software. The box in the mid-
dle shows the steps involved in the clinical evaluation
process. In this work we consider medical device ad-
verse event data as clinical data sources.
To find relevant adverse events to the software in
question text classification can be useful. (Chai et al.,
2013) shows, by using statistical text classification
it is possible to identify health information technol-
ogy incidents from the FDA MAUDE database. The
authors also emphasize using a semi-supervised ma-
chine learning approach to big data analysis of medi-
cal incidents. Apart from the study (Chai et al., 2013),
there is a lack of research that focuses on the concept
of big data and machine learning when it deals with
a large amount of medical device adverse event data
from multiple sources.
The main contributions in this paper are: a) adopt-
ing a modern data ecosystem to deal with the massive
amount of medical device adverse event data, and b)
using text classifiers to classify adverse event data as
per business requirements.
The rest of the paper is organized as follows. Sec-
tion 2 reviewed related works, especially various im-
portant adverse event databases. Based on the re-
view, Section 3 proposes an adverse event data ana-
lytic framework to organize the workflow of the data
analysis for the clinical evaluation procedure. An in-
tegral part of the proposed framework is an adverse
event text classifier. In order to validate the frame-
work, Section 4 conducts experiments to evaluate the
classification accuracy. Finally, Section 5 concludes
the paper and outlines future research directions.
2 RELATED WORKS
The required important regulatory standards for medi-
cal software depend on the country and type of device
in question. In the USA, the Food and Drug Admin-
istration (FDA) is the regulatory authority whereas
in Europe, the European Medicines Agency (EMA)
is the main regulatory authority for medical devices
in Europe. The EMA works closely with the Eu-
ropean Commission, national competent authorities,
and other stakeholders to ensure that medical de-
vices are safe and effective for patients in the EU
and EEA. For medical software some key standards
are (1) ISO 13485: Medical devices – quality man-
agement system (Lie et al., 2020) (2) ISO 14971:
Application of risk management to medical devices
(Flood et al., 2015), (3) IEC 62304: Medical Device
software-system life cycle process (Kim et al., 2019),
(4) IEC 62366: Medical devices- application of us-
ability engineering to medical devices (Costa et al.,
2015), (5) ISO 9001: Quality management systems
(Golas, 2014).
As part of the regulatory standards, medical de-
vice manufacturers use different conformity assess-
ment procedures to demonstrate the performance, ef-
ficacy, and safety of the medical devices. Proving the
safety of medical devices is an ongoing process and
it continues throughout the life cycle of medical de-
vices. Once the device is in the market it starts gen-
erating data regarding safety and performance. Since
medical decisions increasingly depend on data pro-
vided by different medical software, the volume of
data for the safety and performance of medical de-
vices is accordingly increasing over time. For in-
stance, the type of data generated after a medical de-
vice is brought to market may include safety reports,
adverse event reports, and clinical investigations re-
ports. This data can be useful for different conformity
assessment procedures such as clinical evaluation. In
addition to that, this data may contain information
on unknown risks from medical devices. Also, the
post-market adverse event data can be useful, how-
ever, one needs to carefully process the data since the
adverse event reporting system varies among coun-
tries (Kessler, 2007). According to many medical de-
vice regulations, it is medical software manufactur-
ers’ responsibility to generate evidence through dif-
ferent mechanisms mentioned in the regulatory pro-
cess to support the intended purpose of a medical
device in normal use. Clinical evaluation is one of
the processes which can be used to generate evidence
for medical software in both pre-market and post-
market stages. To conduct clinical evaluation pro-
cedures, medical device adverse event data can be
ICT4AWE 2023 - 9th International Conference on Information and Communication Technologies for Ageing Well and e-Health
82
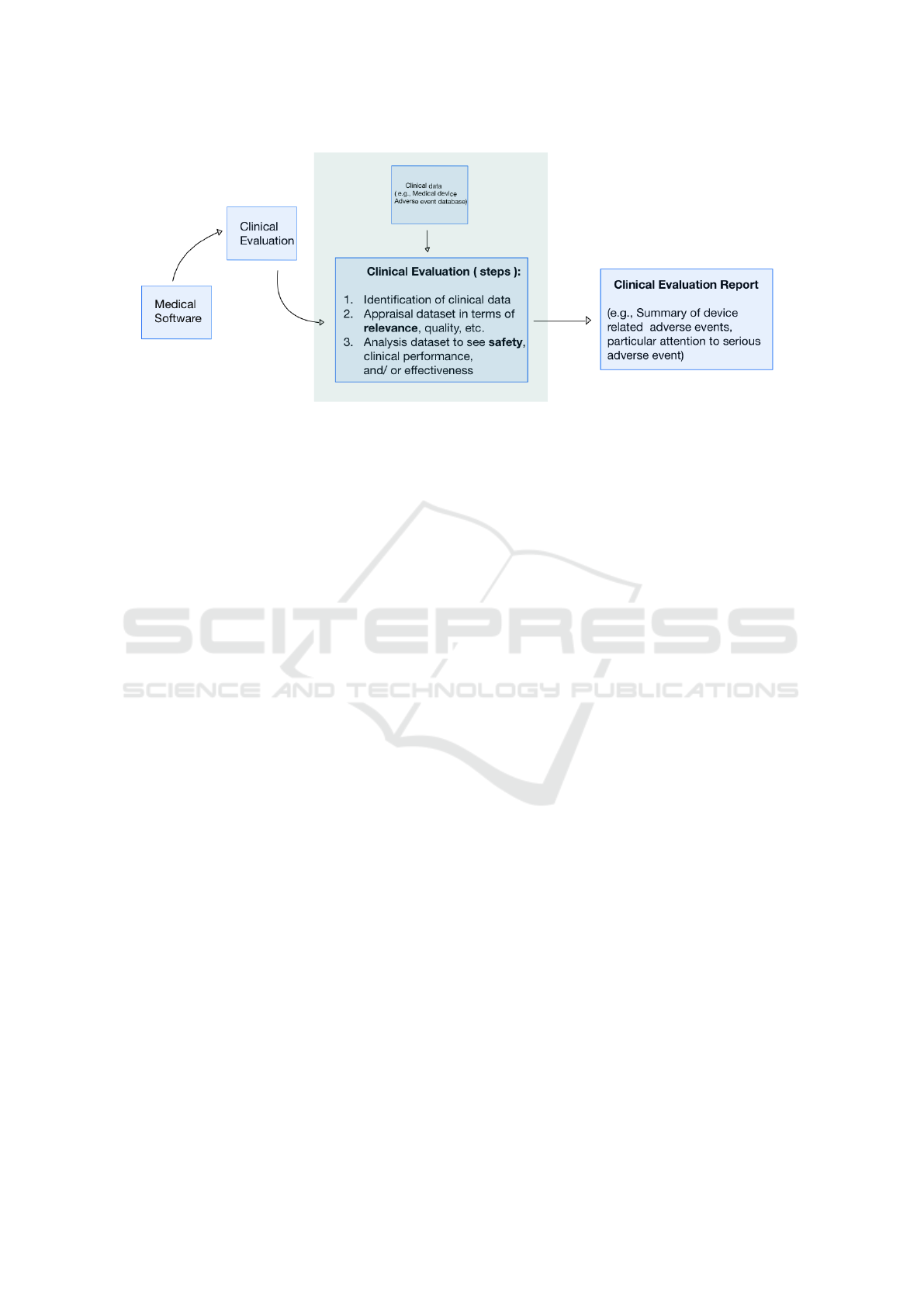
Figure 1: Conceptual Workflow for Conducting Clinical Evaluation for Medical Software.
used(Astapenko, 2019; Commission, 2007; European
Commission, 2020).
This work will focus on collecting clinical data
from desired sources and processing the data to feed
into the clinical evaluation report preparation tasks.
Preparing a clinical evaluation report is out of the
scope of this work. The desired data source may
vary, it depends on the degree of evidence required for
the clinical evaluation report. In this work, adverse
event databases are considered as a desired clinical
data source.
2.1 Adverse Event Databases
Medical device Adverse Event (AE) databases are re-
liable sources for medical software manufacturers to
conduct both pre- and post-market surveillance activ-
ity. Usually, these databases contain adverse event re-
ports submitted by medical device manufacturers, pa-
tients, physicians, and nurses. For example, the FDA
MAUDE database (Manufacturer and User Facility
Device Experience) contains information on medi-
cal device adverse event reports (MDRs) submitted
to FDA by mandatory and voluntary reporters (FDA,
2022). This database represents feedback from differ-
ent stakeholders when a product was brought into the
market. FDA MAUDE represents two kinds of reports
voluntary and mandatory. The mandatory reports are
submitted by manufacturers, importers of medical de-
vices, and device user facilities. The voluntary reports
are submitted by healthcare professionals, patients,
consumers, etc. These reports represent information
on suspected device-associated deaths, injuries, mal-
functions, etc. MAUDE database is publicly available
and provides adverse event data since 1991. These re-
ports support FDA to monitor, and detecting medical
device-related issues and assessing the risk-benefit of
medical devices. The search option on the MAUDE
database has some limitations. (Lisa Garnsey Ensign,
2017) explains how the MAUDE database only shows
a maximum of 500 records and restrict the search
result within the past 10 years for a specific search
query. Due to such restrictions, only a subset of the
available record against a query can be retrieved.
Another database maintained by the FDA is Re-
call. The purpose of the recall database is different
than the MAUDE database. FDA Recall database pro-
vides information on recalled devices to the user of
the device (e.g., Patient, Physician). This database
can be a good source for medical device manufactur-
ers. (Fu et al., 2017) shows that between 2012-2015 a
total of 913 recall data was found related to user inter-
face (UI) software error. To make medical software
safe and effective such information is useful. When
manufacturers acknowledge an issue associated with
their medical device, they usually take either correc-
tive measures or remove the medical device where it
was sold. FDA provides a recall class based on the
degree of risk associated with the medical device in
question. This information may also be important for
the manufacturer to see how serious the event was.
The database is publicly searchable and search results
can be downloaded in CSV format.
Apart from MAUDE and Recall, in the UK
the Medicines and Healthcare products Regulatory
Agency (MHRA) provides data on alerts, recalls, and
safety information about medical devices and drugs.
In Switzerland, the Swiss Agency for Therapeutic
Products (Swissmedic) is a responsible authority to
conduct surveillance on medicines and medical de-
vices available in the Swiss market. In Australia, the
Therapeutic Goods Administration (TGA) is a reg-
ulatory authority that regulates different therapeutic
goods such as medicine, and medical devices avail-
able in the Australian market. All these data sources
can be useful for the clinical evaluation process for
Data Analytics Framework for Identifying Relevant Adverse Events in Medical Software
83

medical software. As these sources contain infor-
mation on medical device safety and effectiveness in
clinical settings.
2.2 Collection of Adverse Event Data
As far as we know, the adverse event data sources
have no uniform layout to collect the desired infor-
mation. Data reside in semi-structured/ unstructured
form on those sources. Some of them provide search
results in downloadable files which contain semi-
structured data, others show the search result on an
HTML table on their the web-page. In this context,
collecting data from dynamic sources and preparing
it for further analysis requires more effort.
Study (Zhang Y, 2019) shows how to collect and
analyze data from FDA Recall database to see user
interface (UI) software error. Authors use keywords
to search data from FDA Recall database, a total of
7,771 records obtained from the database. After that,
data were normalized with two phases to identify po-
tential software errors related to the user interface.
In phase 1, FDC (FDA-determined causes) and key-
words are used to filter out a non-software-related re-
call. Here noted that the FDC information is pro-
vided by the recall database. In phase 2, the au-
thors use a manual approach to analyze the remain-
ing data to identify potential UI-related software er-
rors. During this phase, FDA RES (Recall Enter-
prise System) database was used to identify relevant
information. This study observed, one quarter (25
%) of total identified UI software errors associated
with medical imaging. Although the outcome shows
a well-categorization of UI software errors; however,
the downside is, this work only focused on a single
data source. Especially when it is necessary to col-
lect data from multiple sources and analyze them to
produce clinical evaluation reports, such an approach
could take a long time.
Another study (Fu et al., 2017) shows a slightly
different approach to identifying software-related is-
sues present in medical device recall data. The au-
thors use a set of pre-defined software-related key-
words and a data element named “Reason” which
available on the FDA Recall database. FDA Recall
database provides information on “Reason” of recall.
It is a piece of free text that explains why the medical
device was recalled. Authors use this text data and tag
the textual data with the parts of speech (POS) tagging
technique. POS tagging is a technique that provides a
tag (e.g., noun, adjective, verb, etc.) to each word in
the text/document. Such representation of text is use-
ful and prepares textual data for further analysis (e.g.,
syntactical analysis of text). In the next step, a com-
parison was made between the most relevant nouns
/adjectives and pre-defined software-related keywords
to find the most relevant data. After that, the TF-IDF
algorithm was used to rank the most relevant recall
data.
Moving on to another publication (Chai et al.,
2013) that explores the feasibility of using statistical
text classification to automatically identify health in-
formation technology incidents in the FDA MAUDE
database. This work is a preliminary work that has
shown the use of statistical classification and the po-
tentiality to adopt big data concepts to process a large
amount of adverse event data.
A recent work (Ceross et al., 2021) has shown how
Access Global Unique Device Identification Database
(AccessGUDID) and Global Medical Device Nomen-
clature (GMDN) can be used to analyze medical de-
vice adverse events. The AccessGUDID is a medical
device data database that offers key device identifica-
tion information and is maintained by FDA. On the
other hand, GMDN is a collaborative effort to stan-
dardize terms used in the medical device industry.
Compared with other studies this study has shown a
new way to analyze adverse event data.
Several studies, for example, (Liebel et al., 2020),
also those mentioned previously have shown a com-
mon approach to analyzing adverse event data that is
using relevant keywords. For instance, to find the user
“interface-related” or “software-related” issues they
considered relevant words such as “software”. This
seems to be a reliable approach to mining adverse
event databases.
3 ADVERSE EVENT DATA
ANALYTICS FRAMEWORK
This section will discuss a data analytics framework
and an adverse event text classifier. Based on adverse
event data type and sources we propose a data an-
alytics framework that would support collecting ad-
verse event data, processing it, analyzing it, and fi-
nally preparing data to support clinical evaluation pro-
cedure. In this context, image analysis software is
taken into account that is built on AI technology. As
shown in Figure 2, based on the lessons learned from
(Mac
´
ak et al., 2020), the framework contains 5 stages,
which are data source, data ingestion, data prepara-
tion, intermediate data storage, and data analysis.
3.1 Data Sources
The data source component represents different ad-
verse event data sources useful for medical software.
ICT4AWE 2023 - 9th International Conference on Information and Communication Technologies for Ageing Well and e-Health
84

Figure 2: Adverse event data analytics framework.
Some medical device regulatory guidelines that men-
tioned potential clinical data sources which can be
considered for the clinical evaluation process are-
(1) GHTF/SG5/N7:2012 Clinical Evidence for IVD
medical device – Scientific Validity and Performance
Evaluation (Appendix A) (GHTF, 2012), (2) IMDRF
MDCE WG/N56 FINAL: 2019 Clinical Evaluation –
(Appendix B) (Astapenko, 2019). In addition to that,
the International voluntary group “International Med-
ical Device Regulators Forum (IMDRF) also provides
a list of websites where country-specific medical de-
vice safety information / adverse event data can be
found (IMDRF, 2022). Consulting with those sources
only four adverse event data sources are considered
those are FDA MAUDE, FDA Recall, MHRA, and
SwissMedic. Access Global Unique Device Identifi-
cation Database (GUDID) provides important infor-
mation to identify medical devices. Specifically, this
database provides product-specific FDA code which
is used by other databases such as FDA MAUDE and
Recall. AccessGUDID database is used to find sim-
ilar/comparable products in the first place. Based on
this list FDA MAUDE and Recall database will be
mined. It is worth mentioning that, not all of the cho-
sen data sources provide a uniform layout to access
data. Table 1 shows different options to access data
from desired sources.
3.2 Data Ingestion
As it is shown in Table 1, the targeted data sources
have diverse options to access data. To download the
data from those sources individualized approach has
been taken. For instance, the MAUDE database pro-
vides an option to download data text files whereas
updated data from the Recall database can be ac-
cessed through RSS feed. Both FDA MAUDE and
Recall databases have API (Application Program In-
terface) to access data (U.S. FDA, 2023). In contrast,
data from MHRA and SwissMedic need to be ac-
cessed through RSS (Really Simple Syndication) feed
and web-scraping technology respectively. Thus, a
combination of different approaches needs to be con-
sidered to access targeted data from those sources. It
is important to mention that, in the framework, the
similar or comparable product code list will be re-
viewed and selected by the user manually. To collect
data periodically a scheduling tool can be considered.
After data collection, data need to be stored. The
term “Data Repository” is commonly referred as a
central location where data is collected, organized,
and managed so that it can be used for further busi-
ness operations. The types of data repositories in-
clude databases, data warehouses, and big data stores.
At the time of writing this paper, FDA MAUDE
holds more than 15 million records, and FDA Re-
call holds more than 100 thousand records. The other
two databases, MHRA, SwissMedic hold more than
9 hundred and 9 thousand records respectively. Thus,
managing this large amount of data and bringing them
into a common format is important for analysis.
3.3 Data Preparation
Data integration is important for gaining a unified
view of data. Usually the data integration refers to a
process that combines and transforms data from mul-
tiple sources. As the adverse event data is in semi-
structured or unstructured form ETL/ELT tool can be
chosen to achieve data integration. Both ETL (Ex-
tract, Transform, and Load) and ELT (Extract, Load,
Transform) processes serve the purpose to move data
from source to destination systems and prepare raw
data into an analysis-ready form. The difference be-
tween ETL and ELT is when the loading step would
take place. Compared with ETL, the ELT offer more
flexibility and raw data will be immediately available
to serve other purposes such as exploratory data anal-
ysis.
Data Analytics Framework for Identifying Relevant Adverse Events in Medical Software
85

Table 2 shows, some useful data elements avail-
able on the adverse event databases. On the table, the
FOI TEXT, Reason, Description, and Summary data
fields provide descriptions of the adverse events. To
analyze adverse events these fields are important. Not
all of them have a uniform structure, text length, or
reporting style. This data contains noise like white
space, punctuation, numbers, etc. To feed data into
a machine learning model some pre-processing is re-
quired. As a result, the model could pick the right
signal from the data. Some basic text pre-processing
steps such as removing punctuation, tokenization, and
removing stop words, are used. Based on the use
case, other advanced pre-processing techniques like
stemming, lemmatization, and part-of-speech tagging
(POS) can be considered. After pre-processing, data
needs to be stored in intermediate storage. So that, the
original data will remain intact.
3.4 Intermediate Data Storage
The next component in Figure 2 is intermediate data
storage. Intermediate data storage is considered in the
pipeline to keep the original raw data intact. Adverse
event text data may contain noise, such as punctua-
tion, stop words, unwanted characters, etc. So this
data needs to be cleaned before going for further anal-
ysis. After pre-processing data is transformed and the
step is irreversible. If we operate on raw data there
is no way to go back, once data is transformed. So it
would be a good idea to keep raw data as it is and con-
sider saving transformed data in a separate location.
The intermediate storage can be thought of as a data
lake where data will write once and read many times
for analytic purposes. Another advantage of hav-
ing intermediate storage is cleaned and pre-processed
data can be used for other analytical purposes with-
out doing repetitive pre-processing. Different file for-
mats can be considered such as Parquet, or Pickle to
achieve space-efficient storage.
3.5 Data Analysis
Turning now to the data analysis component which
contains the machine learning model to classify ad-
verse event data. The model would take pre-processed
data from the previous step. Then it will predict each
adverse event record whether it is relevant for an im-
age analysis software or not and finally provide the
output in an excel format. Building a text classifier
requires some labeled data set. To the best of our
knowledge, no publicly available labeled data set is
available to build a text classifier on medical device
adverse events. Thus, we need to prepare some data
sets with specific labels. In this work, we use two la-
bels, those are “Relevant” and “Not relevant”. The
“Relevant” label represents an adverse event record
that is relevant to image analysis software. On the
other hand, the “Not relevant” label represents an ad-
verse event record that is not relevant for image anal-
ysis software. To build an adverse event text classi-
fier supervised machine learning approach is consid-
ered. Two different supervised machine learning al-
gorithms with different vectorization approaches are
tested. The algorithms include Support Vector Ma-
chine (SVM) and Random Forest Classifier and the
vectorization techniques are TF-IDF and word2vec.
4 EVALUATION
This section represents the outcome of applying com-
monly used matrices to find which algorithm per-
forms well to classify adverse events text. The evalu-
ation matrices are - Accuracy, Precision, Recall, and
Cross-validation.
4.1 Evaluation Setting
The accuracy is compared with a simple baseline. The
goal is to validate whether the trained model works
better than the baseline. We use DummyClassifier
class from scikit-learn. It provides strategies like
“most frequent” where the method returns the most
frequent class label. We applied this for LinearSVC
model only.
The whole labeled data set is split into 80-20 ratio.
Twenty percent data set is used as a test set. For cross-
validation, the whole data set is considered and split
into k-subsets (k=9) and the holdout method is re-
peated k times. Each time, one of the k-subsets is used
as the test set and other k-1 subsets are put together
to be used to train the model. Cross-validation allow
to use the whole data set and it helps the model to
learn on more data. As it is shown in Table 3 and Ta-
ble 4, the Support Vector Machine (SVM) algorithm
performs well in different matrices. Only LinearSVC
with TF-IDF model is considered for cross-validation.
In the confusion matrix, the “Relevant” class is
considered as true positive, and the “Not relevant”
class is considered as true negative. Precision repre-
sents what proportion of predicted positive is positive
or how good the classification model is at predicting
the ”relevant” class. The recall represents what pro-
portion of real positive values are identified by the
classification model.
ICT4AWE 2023 - 9th International Conference on Information and Communication Technologies for Ageing Well and e-Health
86
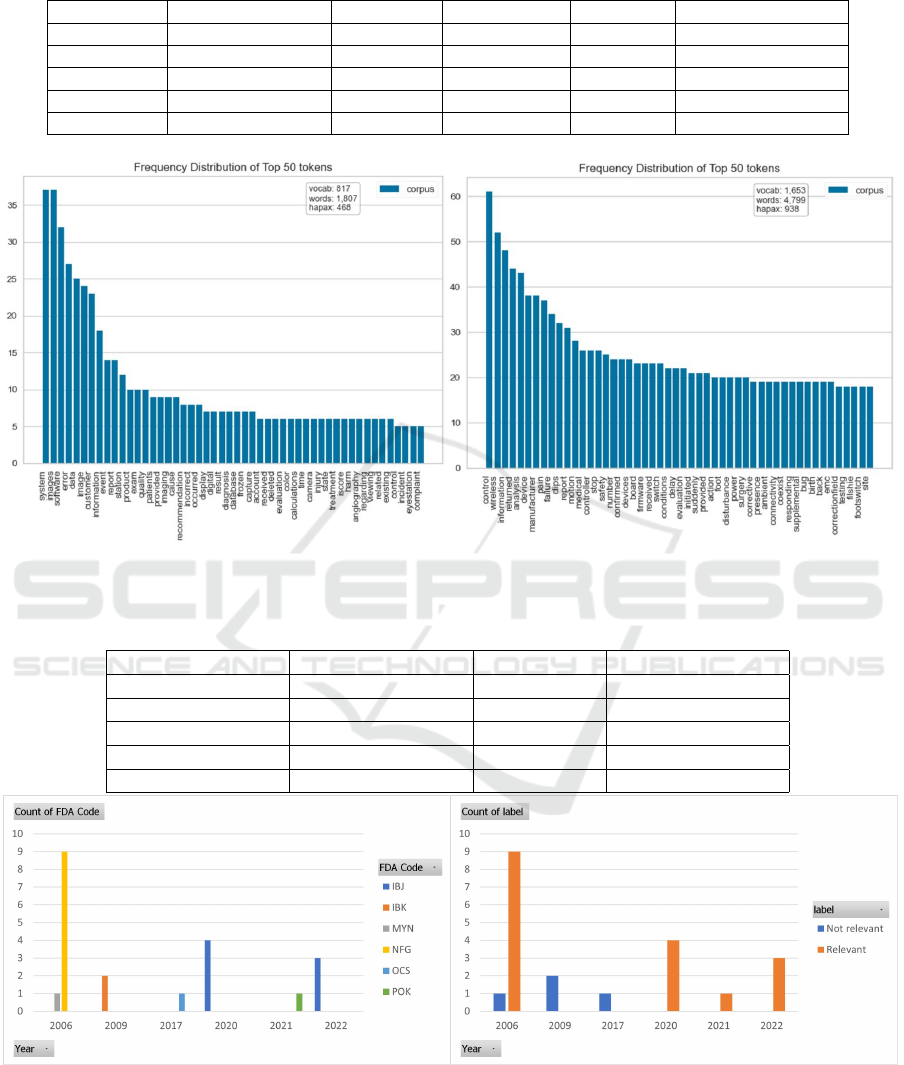
Table 1: Possible Ways to Access Data from Targeted Sources.
Website URL Generation Robots.txt Sitemap.xml RSS Feed Downloadable file
MAUDE x x - - x
Recall x x - x x
MHRA x - x x -
SwissMedic x - - - x
GUDID - - - x x
(a) Relevant (b) Not Relevant
Figure 3: Visualizing feature for both classes.
Table 2: Useful Data Elements Available on Adverse Event Databases.
FDA MAUDE FDA Recall SwissMedic MHRA
Manufacturer Product Description Manufacturer Title
Brand Name Recall Class Date Issue Date
Date Received Recall Posting Date Brand Name Alert Type
Adverse Event Flag Manufacturer Description Medical Specialism
FOI TEXT Reason - Summary
(a) Favorable adverse event records to image analysis software(b) Classification of adverse event data using the trained model
Figure 4: Use trained model on new adverse event data. Database: FDA MAUDE, Timeframe:(2000-2022).
Data Analytics Framework for Identifying Relevant Adverse Events in Medical Software
87

4.2 Training Data
To the best of our knowledge, there were no exist-
ing publicly available datasets to train a model for ad-
verse event data. Thus, data annotation has to choose
to create some labeled data. Data annotation requires
time and strong domain knowledge. There are sev-
eral approaches that can be followed to annotate data.
When no training data is available, manual annotation
or bootstrapping can be chosen. Manual annotation is
a time-consuming process. Also, it is ideal to have
3-5 annotators, who would work on the same dataset
and annotate them individually. Later the annotated
data will compare, if any disagreement occurs on any
specific record, it should be further analyzed.
We have labeled 162 data with two classes. It is
worth mentioning that all 162 records come from the
MAUDE database. The reason is, during exploratory
data analysis it was observed this database and also
Recall database follow a structure in their reporting
and explain the reason for the event. This is useful to
capture intuition on adverse events. Figure 3 visual-
izes top-frequent terms in the labeled data. Here the
label ”relevant” means, the adverse event relevant to
image analysis software and ”not relevant” means the
event is not related to image analysis software.
We take our 162 labeled data and count the word
frequency, where the bag-of-words (BOW) model is
used. The BOW model takes each adverse event de-
scription text and treats it as a bag of words, where the
order of words is ignored. The adverse event termi-
nology working group led by IMDRF, is working on
improving, harmonizing, and standardizing terminol-
ogy as regards medical device adverse event (IMDRF,
2019). To find the most informative terms within la-
beled data this resource was used. Furthermore, the
technical report titled “IEC/TR 80002-1:2009 Medi-
cal device software — Part 1: Guidance on the ap-
plication of ISO 14971 to medical device software”
includes “Annex B”, which provides additional guid-
ance on software function areas and hazards(IEC,
2009) also used to find the most informative terms.
It is worth mentioning that only adverse event/hazard
definitions associated with image analysis software
were considered.
4.3 Evaluation Results
We achieved 78 percent accuracy for the dummy clas-
sifier. The result of the dummy classifier indicates that
the trained LinearSVC model adds some value. Table
3, lists the accuracy scores for different model combi-
nations. The LinearSVC shows the highest accuracy
(96 percent) compared with other models. In Table 4
shows, precision and recall scores for different model
combinations. For precision and recall the best score
was achieved with the SVM algorithm that was uti-
lized with the TF-IDF model, employing a linear ker-
nel function. In this case, for the “Relevant” class
precision and recall were 1 and 0.89 respectively. In
the test set, the total observation was 33, among them,
9 were “Relevant” class. The trade-off between preci-
sion and recall depends on the business use case. For
instance, if the cost of not identifying an adverse event
is high then we should consider improving the recall
score. With the current recall score of 0.89, the model
is able to capture most of the adverse event text that
is relevant to image analysis software. Comparing the
score with other kernel functions, it seems that the
adverse event text data is linearly separable. Finally,
cross-validation was applied to the same model. We
achieved a 0.94 (mean) score where k=9. This score
is close to what we achieved for other matrices. Thus,
this score can be taken as the model’s true potential.
Figure 4 shows an output of the proposed data an-
alytics framework. The illustration represents the data
based on the FDA MAUDE database and the trained
model applied to new adverse event data. Between
2002-2022, a total of 21 adverse events have been
identified. Among them, 17 records were assigned to
a relevant class and 4 were assigned to a not-relevant
class by our model. The similar/comparable FDA
product code included IBJ, IBK, MYC, MYN, NFG,
OCS, OEB, POK, QBS, QDQ, QJU, QNP. Based on
these product codes a total of 35 records are stored in
intermediate storage. To use the model on new data,
we have taken those records that contain more than 50
words in the adverse event description.
4.4 Discussions
We found that the AccessGUDID, MAUDE, and Re-
call databases can be connected by using the FDA
product code. This linkage is useful to find a specific
subset of data from the adverse database. First, it is
important to identify the most similar or comparable
product code from AccessGUDID database to the de-
vice in question. Later the selected product code can
be used to retrieve data from the other two databases
MAUDE and Recall. During our data analysis, it was
observed between 2000-2022 only MAUDE database
contains more than fifteen million records and the Re-
call database contains more than one hundred thou-
sand records.
By using specific product codes, it is possible to
take a subset of data that represents adverse event data
useful for specific medical products. Finding sim-
ilar/comparable product not only helps to mine ad-
ICT4AWE 2023 - 9th International Conference on Information and Communication Technologies for Ageing Well and e-Health
88

Table 3: Accuracy.
Algorithm Vectorization technique Accuracy Note
SVM
TF-IDF 0.96 LinearSVC
0.79 Polynomial, Degree 3
0.87 RBF, Degree 3
word2vec 0.67 LinearSVC
0.67 Polynomial, Degree 3
Random Forest
TF-IDF 0.88 -
word2vec 0.88 -
Table 4: Precision and Recall.
Algorithm Vectorization Precision Recall Class Observation Note
SVM
TF-IDF 1 0.89 Relevant 9 Linear
0.96 1 Not relevant 24
1 0.22 Relevant 9 Polynomial, Degree 3
0.77 1 Not relevant 24
1 0.56 Relevant 9 RBF, Degree 3
0.86 1 Not relevant 24
word2vec 0.68 1 Not relevant 22
0 0 Relevant 11 LinearSVC
0.68 1 Not relevant 22 Polynomial, Degree 3
0 0 Relevant 11
Random Forest
TF-IDF 1 0.64 Relevant 9 -
0.85 1 Not relevant 24 -
word2vec 0.85 1 Not relevant 22 -
1 0.64 Relevant 11 -
verse event data but this knowledge can also be used
for other purposes as well. For instance, market anal-
ysis, product differentiation, etc. This can be achieved
by analyzing the AccessGUDID database. During
the adverse event data analysis we dealt with large
amounts of text data. It requires more computational
power and resources. Long processing time was a key
challenge. A multi-processing tool like Spark can be
useful to resolve this problem.
5 CONCLUSIONS
This paper has proposed a data analytics ecosystem to
analyze medical device adverse event reports for clin-
ical evaluations. The proposed framework consists
of collecting various data sources, via data ingestion,
preparation, and data staging, and finally data analy-
sis. We found that one of the important components
in the framework is the adverse event text classifier.
In order to validate the framework and adverse event
text classifier, we have conducted an experiment to
evaluate the accuracy of different machine learning
algorithms.
The experimental result has shown that an adverse
event text classifier can be an integral part of this
ecosystem and it is feasible to achieve solid classifica-
tion accuracy. Thus, the adverse event text classifier
and the framework can support other medical devices’
regulatory tasks such as active post-market surveil-
lance activity. Also, this work has indicated a new
research stream that is using knowledge from the data
science domain to deal with different medical device
regulatory affairs tasks and clinical evaluation tasks.
As future work we will focus on building an ad-
verse event text classifier that would classify adverse
event text at a more granular level. For example, what
semantically the adverse event text means, and how
to represent an issue related to a “software execution”
or an “image orientation” related issue.
REFERENCES
Agyei, E. E. Y. F., Pohjolainen, S., and Oinas-Kukkonen,
H. (2022). Impact of medical device regulation on de-
veloping health behavior change support systems. In
Baghaei, N., Vassileva, J., Ali, R., and Oyibo, K., edi-
tors, Persuasive Technology - 17th International Con-
ference, PERSUASIVE 2022, Virtual Event, March
29-31, 2022, Proceedings, volume 13213 of Lecture
Notes in Computer Science, pages 1–15. Springer.
Astapenko, E. M. (2019). Clinical evaluation. International
Medical Device Regulators Forum. IMDRF MDCG
WG/N56FINAL.
Data Analytics Framework for Identifying Relevant Adverse Events in Medical Software
89

Ceross, A., Bergmann, J., et al. (2021). Tracking the
presence of software as a medical device in us food
and drug administration databases: Retrospective data
analysis. JMIR Biomedical Engineering, 6(4):e20652.
Chai, K., Anthony, S., Coiera, E., and Magrabi, F. (2013).
Using statistical text classification to identify health
information technology incidents. Journal of the
American Medical Informatics Association : JAMIA,
20.
Chen, J. H., Alagappan, M., Goldstein, M. K., Asch, S. M.,
and Altman, R. B. (2017). Decaying relevance of clin-
ical data towards future decisions in data-driven inpa-
tient clinical order sets. Int. J. Medical Informatics,
102:71–79.
Commission, E. (2007). Clinical Evaluation: A Guide for
Manufacturers and Notified Bodies under Directives
93/42/EEC and 90/385/EEC. European Commission,
Brussels, Belgium.
Costa, S. B. D., Beuscart-Z
´
ephir, M., Bastien, J. M. C.,
and Pelayo, S. (2015). Usability and safety of soft-
ware medical devices: Need for multidisciplinary ex-
pertise to apply the IEC 62366: 2007. In Sarkar, I. N.,
Georgiou, A., and de Azevedo Marques, P. M., edi-
tors, MEDINFO 2015: eHealth-enabled Health - Pro-
ceedings of the 15th World Congress on Health and
Biomedical Informatics, S
˜
ao Paulo, Brazil, 19-23 Au-
gust 2015, volume 216 of Studies in Health Technol-
ogy and Informatics, pages 353–357. IOS Press.
European Commission (2020). Guidance on clinical evalu-
ation (mdr) / performance evaluation (ivdr) of medical
device software. [Accessed: March 11, 2023].
European Parliament and Council of the European Union
(2017). Regulation (eu) 2017/745 of the european par-
liament and of the council of 5 april 2017 on medical
devices.
FDA (2022). Maude - manufacturer and user facility device
experience. https://www.accessdata.fda.gov/scripts
/cdrh/cfdocs/cfmaude/search.cfm.
Flood, D., McCaffery, F., Casey, V., McKeever, R., and
Rust, P. (2015). A roadmap to ISO 14971 implemen-
tation. J. Softw. Evol. Process., 27(5):319–336.
Fu, Z., Guo, C., Zhang, Z., Ren, S., Jiang, Y., and Sha,
L. (2017). Study of software-related causes in the
fda medical device recalls. In 2017 22nd Interna-
tional Conference on Engineering of Complex Com-
puter Systems (ICECCS), pages 60–69.
GHTF (2012). Clinical evidence for ivd medical devices
– scientific validity determination and performance
evaluation. Global Harmonization Task Force, Study
Group 5 Final Document GHTF/SG5/N7:2012.
Golas, H. (2014). Risk management as part of the
quality management system according to ISO 9001.
In Stephanidis, C., editor, HCI International 2014
- Posters’ Extended Abstracts - International Con-
ference, HCI International 2014, Heraklion, Crete,
Greece, June 22-27, 2014. Proceedings, Part II, vol-
ume 435 of Communications in Computer and Infor-
mation Science, pages 519–524. Springer.
IEC (2009). Medical device software — part 1: Guidance
on the application of iso 14971 to medical device soft-
ware. Technical report.
IMDRF (2019). Medical device problem codes. Accessed
on: March 12, 2023. URL: https://www.imdrf.
org/working-groups/adverse-event-terminology/
annex-medical-device-problem.
IMDRF (2022). Imdrf -safety information.
https://www.imdrf.org/safety-information.
Kessler, L. (2007). Ghtf sg5: Clinical evaluation.
Kim, D., Lee, B., and Lee, J. (2019). Building a rule-based
goal-model from the IEC 62304 standard for medi-
cal device software. KSII Trans. Internet Inf. Syst.,
13(8):4174–4190.
Leveson, N. (1995). Medical devices: The Therac-25. IEEE
Computer, 26(7):18–41.
Lie, M. F., S
´
anchez-Gord
´
on, M., and Palacios, R. C. (2020).
Devops in an ISO 13485 regulated environment: A
multivocal literature review. CoRR, abs/2007.11295.
Liebel, T. C., Daugherty, T., Kirsch, A., Omar, S. A., and
Feuerstein, T. (2020). Analysis: Using the fda maude
and medical device recall databases to design better
devices. Biomedical Instrumentation & Technology,
54(3):178–188.
Lisa Garnsey Ensign, K. B. C. (2017). A primer to the struc-
ture, content and linkage of the fda’s manufacturer and
user facility device experience (maude) files. Egems
(generating Evidence & Methods to Improve Patient
Outcomes), 5(1).
Mac
´
ak, M., Ge, M., and Buhnova, B. (2020). A
cross-domain comparative study of big data architec-
tures. Int. J. Cooperative Inf. Syst., 29(4):2030001:1–
2030001:27.
Nadakinamani, R. G., Reyana, A., Kautish, S., Vibith,
A. S., Gupta, Y., Abdelwahab, S. F., and Mohamed,
A. W. (2022). Clinical data analysis for prediction of
cardiovascular disease using machine learning tech-
niques. Comput. Intell. Neurosci., 2022:2973324:1–
2973324:13.
U.S. FDA (2023). Openfda. https://open.fda.gov/. Accessed
March 11, 2023.
Zhang Y, Masci P, J. P. T. H. (2019). Research: User inter-
face software errors in medical devices: Study of u.s.
recall data. Biomedical Instrumentation & Technol-
ogy, 53(3).
ICT4AWE 2023 - 9th International Conference on Information and Communication Technologies for Ageing Well and e-Health
90
