
Ongoing Work to Study the Underlying Statistical Patterns of
Oesophageal Chromothripsis
Jack Fraser-Govil and Zemin Ning
The Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton, Cambridge CB10 1SA, U.K.
Keywords:
Chromothripsis, Bayesian Statistics, Chromosome Rearrangement.
Abstract:
In this position paper we demonstrate our ongoing efforts to develop and test a number of statistical tools
and methedologies which allow us to study the underlying statistical properties of a genetic sequence which
has undergone chromothripsis, and hence provide some novel probes into the mechanisms which cause such
catastrophic genomic rearrangement. Using these tools, we study an oesophogeal cancer sample showing more
than 1000 rearrangements, with 800 of these on chromosome 6. By studying this chromosome, we challenge
a prevalent idea within the literature: that chromothripsis breakpoints are non-random, finding instead that
despite a high degree of clustering, the clusters themselves are uniformly distributed across the chromosome.
We also show that although 3-dimensional proximity is a tempting explanation for the rearrangement pattern,
the statistical evidence does not favour it at the current time. In addition, we attempt to disambiguate some of
the terminology surrounding chromothripsis.
1 INTRODUCTION
The conventional model of cancer development posits
that the inciting genetic defects are the result a grad-
ual accumulation of point mutations and rearrange-
ments, eventually resulting in the activation of onco-
genes. The discovery of chromothripsis (Stephens
et al., 2011), however, presented a potential alterna-
tive pathway: that of a genetic crisis resulting in a
massive genomic rearrangement in a single event.
The chromothripsis phenomomenon was charac-
terised by a number of ‘breakpoints’ which showed
an unusual level of clustering, and an oscillation in the
copy number variation which seemed to indicate that
the genome had been ‘shattered’ into multiple distinct
fragments, before a DNA repair mechanism had erro-
neously repaired these broken links into a contiguous
but now cancer-causing sequence.
The view that chromothripsis is the result of a sin-
gle catastrophic event has, however, been challenged
(Solorzano et al., 2013). This is complicated further
by that fact that some (i.e. (Korbel and Campbell,
2013)) use simultaneity as an axiomatic part of their
definition of chromothripsis - which in turn precludes
the study of any evidence of chromothripsis as an ex-
tended process. We therefore emphasise throughout
this work the importance of using a constent termi-
nology, which we robustly define in section 1.1.
The actual underlying mechanics of chromothrip-
sis, whether they be instantaneous or sequential, or
even if multiple such pathways exist, remain an open
question. The aim of our ongoing work is to use sta-
tistical tools to attempt to gain insight into the ways
in which the breakage and repair processes imprint
themselves onto the resulting cancer genome, using
a particularly prominent oesophogeal cancer as our
testbed for these tools.
In doing so, we introduce a Bayesian inference
engine (which will be published as its own separate
work, (Fraser-Govil, 2023)), and discuss our ongoing
work to use this tool to study the break process using
the CHROMOSPA tool, and then finally leveraging the
statistical engine to identify if the repair process can
be associated to spatial proximity within the nucleus,
using HiC data.
This is ongoing work, so our conclusions and data
are provisional for the moment, but we hope that this
elucidates the direction and motivations of our re-
search.
1.1 Terminology
As noted, the terminology surrounding chromothrip-
sis has developed and shifted since its discovery, with
some explanatory features present in the original stud-
ies since being used as definitional elements. It is
therefore vital when discussing chromothripsis that
one is careful to define exactly what one means by
that term.
In our work, we emphasise that chromothripsis
276
Fraser-Govil, J. and Ning, Z.
Ongoing Work to Study the Underlying Statistical Patterns of Oesophageal Chromothripsis.
DOI: 10.5220/0011785600003414
In Proceedings of the 16th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2023) - Volume 3: BIOINFORMATICS, pages 276-283
ISBN: 978-989-758-631-6; ISSN: 2184-4305
Copyright
c
2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)

Ostracon
Chromothripsis
Hologenome
Mignogenome
Figure 1: A Diagram indicating our chosen terminology for
chromothripsis.
is a phenomenological term, describing an observed
pattern in the data. Formally speaking, we define
chromothripsis as a process which generates a large-
scale genomic rearrangement which possesses statis-
tical indiators (i.e. copy number oscillations, cluster-
ing) which lie in tension with the standard, sequential
mechanism of cancer evolution.
This definition is intentionally ambivalent to the
precise mechanism by which chromothripsis oper-
ates - either by the original ‘shattering’ model, or
by some extended process which nevertheless oper-
ates distinctly from the previously understood mech-
anisms of cancer formation.
In keeping with our efforts to use clear terminol-
ogy, we also provide the following definitions to al-
low us to unambiguously distinguish between the key
components of chromothripsis:
• Hologenome (also Holosequence etc.): The origi-
nal, unbroken genetic sequence
• Mignogenome (also Mignosequence etc.), the se-
quence which has been drastically rearranged by
the process of chromothripsis.
• Ostracon, an individual unbroken, contiguous,
segment of the hologenome present within the
mignogenome (from ὄστρακον, broken fragment
of pottery with letters inscribed)
• Breaks (also breakpoints), the points on the
Hologenome which form the original edges of
the ostracons, specified through a single coordi-
nate: that of the chromosomal coordinate in the
hologenome.
• Joins (also joinpoints), the points on the
Mignogenome which form the edges of adjoining
ostracons in the mignogenome
Under this terminology, therefore, chromothripsis
is the generic name given to any process which gener-
ates a mignogenome, either by shattering the genome
into ’ostracons’ which are then reassembled, or by a
more extended process which simply mimics this be-
haviour.
We emphasise that there is a distinct and important
difference between the locations of ostracons within
the hologenome and their location in mignogenome,
and that there are potentially two different driving
forces behind them.
The location of ostracons (or, more precisely,
the edges of the ostracons - the breaks) within the
hologenome are a result of the destructive process:
the process broke the hologenome at several loca-
tions, and patterns in the individual positions of the
breakpoints are characteristic of this process.
The location of ostracon pairs within the
mignogenome, however, is indicative of the repair
process - the process which repaired the genome after
the shattering event. Patterns in which pairs of ostra-
cons are adjacent are indicative of how this process
occured.
We have no particular a priori reason to assert that
these processes are related, and thus we should study
them as distinct - though sequential - processes.
2 DATA
2.1 Identification of Breakpoints
We used STEPPINGSTONE
1
to parse the reads of
the oesophogeal cancer samples, and extract a list
of identified breakpoints. STEPPINGSTONE func-
tions by noting that when reads originating from a
mignogenome are aligned to a reference, the edges of
the ostacons will appear as chimeric reads – with the
sequences on either side of the joinpoint aligning to
different parts of the genome – even though they are
genuinely contiguous sequences in the mignogenome.
STEPPINGSTONE reports these chimeric points
via the two chromosomal coordinates within the
hologenome that the chimeric reads correspond to.
Work is ongoing to fully assemble this information
to generate the full mignogenome sequence.
2.2 Test Sample
For the current work-in-progress demosntrated in this
work, we use the sequencing data of an oesophogeal
cancer sample (Sanger sequencing ID OSEO-103).
This is a rather remarkable sample due to the sheer
number of breakpoints: more than 1,000 breakpoints
identified with more than 5 reads confirming them.
The majority of the high-coverage breakpoints are
confined to Chromosomes 6 and 9. For our work here,
we focus on chromosome 6, which contains more than
800 breakpoints with more than 10 reads confirming
them.
1
https://github.com/wtsi-hpag/steppingStone
Ongoing Work to Study the Underlying Statistical Patterns of Oesophageal Chromothripsis
277
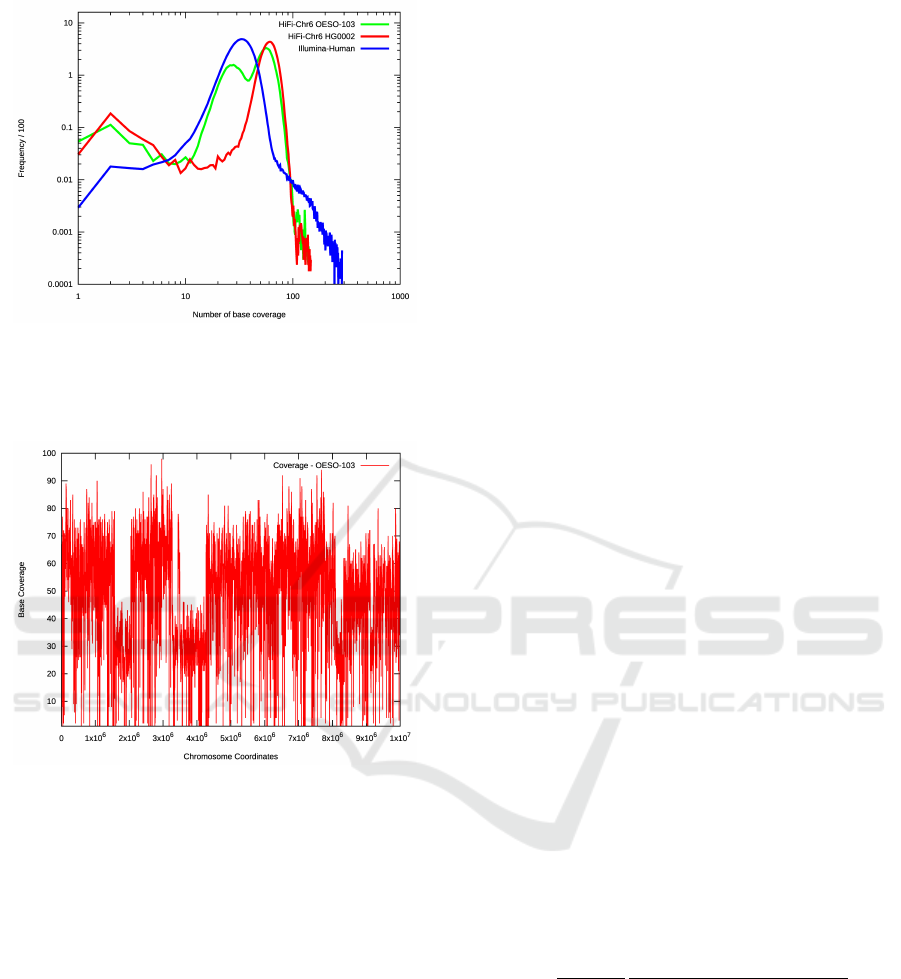
Figure 2: The global coverage distribution of chromosome
6 in the OESO-103 sample, demonstrating a bimodal distri-
bution, with one peak at apporximately half the coverage of
the other, consistent with a change occuring on only one of
a homologous pair.
Figure 3: A snapshot of the per-base coverage of chromo-
some 6 in the OESO-103 sample, showing a marked drop
in coverage around the site of identified breaks at ∼ 2 ×10
6
and ∼ 4 × 10
6
: approximately a drop of one-half, indicat-
ing the presence of two haplotypes generated by one of the
diploid pair having undergone a break/joining at this loca-
tion.
Due to the unusual nature of this particular sam-
ple, it is relatively unambiguous that it is the result of
a chromothripsis process, though we note from Fig-
ures 2 and 3 that there are clear signs from the cov-
erage data that multiple hapolotypes have been gener-
ated - samples which are less extreme might have to
demonstrate more robustly that they are genuinely in
tension with traditional models of cancer formation.
2.3 Future Data
It is evident that the statistical importance of our re-
sults is limited until we can demonstrate that they
hold true across multiple instances of chromothripsis,
rather than just the single case we currently possess.
It is of vital importanance for our future work that
we test these tools on additional chromothripsis sam-
ples. However, as this simply serves as a proof of
concept for the information it is possible to extract,
we continue with our single sample until more data is
available.
3 HYPOTHESISTESTER TOOL
In order to robustly examine models for explaining
the data extracted from our chromothrispis sample,
we must have a statistically robust mechanism for as-
sessing which models are better in explaining the fea-
tures of the data.
We have found the standard statistical tools such
as statistical significance testing generally unsuitable
for this task, and thus have joined the chorus (i.e.
(Stang et al., 2010)) of those advocating a Bayesian
approach to model testing and selection.
The primary concern is that, in general, a more
flexible model (i.e. one with more free parameters
which can be fit to the data) will always be able to
provide a better fit than a model with fewer param-
eters, thus, complexity is favoured over simplicity.
As a pathological example, it is always possible to
draw a perfect polynomial fit to N datapoints if the
polynomial is of N − 1
th
order. If one is faced with
choosing between a straight line which close to (but
not exactly through) 80 data points, or one which
contains terms of order x
79
but which perfectly goes
through every datapoint, any method which relies
purely on goodness-of-fit would choose the highest-
dimensional model, no matter how ludicrous those
are.
Bayesian tools, however, allow us to directly ac-
cess the relative likelihood between two models, A
and B in explaining the data D, in the form of the
odds-ratio:
R
AB
=
Prior(A)
Prior(B)
R
d
⃗
λProb
A|D,
⃗
λ
Prior(
⃗
λ)
R
d⃗µProb(B|D,⃗µ) Prior(⃗µ)
(1)
Here the Prior is the initial belief we have in the
model (and its parameters, µ and λ). If R
AB
≫ 1, then
hypothesis A is much more likely to be true than hy-
pothesis B. Of course, more data might alter this con-
clusion, and Hypothesis C might be better still, but
this provides an objective, numerical way to assess
which model is best.
Although the underlying theory for computing
these odds ratios is available in most introductory
Bayesian Statistics textbooks, the techniques are often
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
278
only easily applicable in pathological, simple exam-
ples, and there is remarkably little computational sup-
port enabling widespread use in the non-pathological
cases. To this end, we have developed the flexible
and easy-to-use Bayesian Hypothesis Testing Engine
- HYPOTHESISTESTER - available in both C++ and
Python implementations, which we hope will make
computing odds ratios simple and robust for a wider
audience.
The underlying mechanics of the HYPOTHE-
SISTESTER work and its associated optimisation rou-
tine, AHAB, will be published as (Fraser-Govil and
Boubert, 2023) and (Fraser-Govil, 2023).
4 CHROMOSPA: SIZE AND
POSITION ANALYSIS
Several works (Stephens et al., 2011; Maher and Wil-
son, 2012; Rausch et al., 2012) have noted that the
identified breakpoints in chromothripsis show signif-
icant clustering - however, this clustering was iden-
tified as a signal of a “non-random distribution”, a
claim which has since been repeated elsewhere in the
literature (Righolt and Mai, 2012; Korbel and Camp-
bell, 2013; Mardin et al., 2015; Voronina et al., 2020).
However, we note that clustering is emphatically not
a signal of “Non-randomness”, which would imply a
mechanistic, exactly predictable pattern to the break-
points, for which significant evidence has not been
demonstrated. Clustering should instead be seen as an
indicator of a bias in the underlying probability dis-
tribution - we must instead interpret the prior use of
“non-random” instead to mean non-globally-uniform.
The location of the breakpoints can give insight
into the underlying distributions which caused the
fracturing of the genome. To this end, we are devel-
oping the CHROMOSPA tool
2
, which performs statis-
tical analysis on the locations of breakpoints and the
size of the resulting ostracons. This is still a work
in progress, however we discuss briefly some of our
preliminary results.
4.1 Ostracon Size
Once a list of breakpoints has been inferred via STEP-
PINGSTONE, the length of each ostracon can be in-
ferred simply by subtracting successive breakpoint in-
dices from each other: if two neighbouring break-
points on a chromosome are found at i and j respec-
tively, the length of the ostracton is |i − j |.
2
https://github.com/wtsi-hpag/chromoSPA
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
10 10 10 10 10 10 10 10 10
Fractional Fragment Length
Chrom6 observed
Uniform Probability Fit
Figure 4: The observed frequency of ostracon sizes (given
as a fraction of the entire chromosome 6 length) on Chro-
mosome 6 of our sample.
We note that this inference of the ostracon length
assumes that chromothripsis occurs only on a single
copy of (in this case) chromosome 6, since STEP-
PINGSTONE is unable to phase the reads, and hence
cannot distinguish between breakpoints occuring on
different homologous chromosomes. We justify this
by noting in Figs. 2 and 3 that the drop in coverage
of ≈ 50% supports the notion that only one copy of
the chromosome is affected by chromothripsis. How-
ever, future work in this area should make the statis-
tical inference robust against the possibility of multi-
homolog chromothripsis.
Figure 4 shows the observed distribution of the
breakpoints, along with the best-fit probability model,
assuming that breakpoints occur uniformly across the
chromosome. As expected, we see a clear bias of
more smaller ostracons than the uniform model would
predict: this is due to the previously identified clus-
tering of breakpoints, which produces many smaller
ostracons due to the close proximity of the breaks.
The pattern in Fig. 4 is therefore a superposition
of the length of ostracons within clusters and of the
distance between clusters.
Our analysis shows that the distances between os-
tracons are well explained by a Gaussian mixture
model, such that the probability of a break occuring
at position x is given by:
p(x) =
∑
i
w
i
N
i
exp
−
(x − µ
i
)
2
2σ
2
i
(2)
That is, the probability distribution of each cluster is
(approximately) Gaussian, with σ
i
≈ 10kb, which re-
sults in osctacon lengths which are in turn distributed
in a Gaussian fashion, with size 5 ± 2kb.
Interestingly, however, we find that the distribu-
tion of the focal points of the clusters - the µ
i
values
Ongoing Work to Study the Underlying Statistical Patterns of Oesophageal Chromothripsis
279
- shows no signifiant bias. This is surprising as we
have already identified several supra-chromosomal
patterns (significant chromothripsis only occurs on 1
copy of chromosomes 6 and 9, for example), how-
ever, it seems of those chromosomes which do suffer
chromothripsis, the process results in cluster hotspots
which have no particular positional bias in the chro-
mosome.
This is a tantalising hint that, although the breaks
are highly clustered around the focal points, the dis-
tribution of the focal points is highly random and uni-
form within a chromosome; subject to the chromsome
being a chromothripsis candidate in the first place – a
seemingly odd, random process amidst an otherwise
highly ordered heirarchy of events.
We do note, however, that we are limited by our
single chromothripsis sample: comparisons with mul-
tiple samples might reveal that the same positions oc-
cur in multiple events, which would indicate that there
is something special about these locations, but that
this special property is uniformly distributed in the
chromosome.
5 ContactPoint ANALYSIS
In this section we turn to analysing the joinpoints
generated by chromothripsis: studying why a given
ostracon ends up joined to another in the final
mignogenome.
One plausible hypothesis is that, after a breakage
occurs, the DNA strands are repaired on the basis of
proximity: once a breakpoint forms, generating an
ostracon with a free end, the joinpoint then prefer-
entially occurs between ostracons which are spatially
close together. Since DNA within the cell forms a
complex 3D structure, the resulting joinpoints when
projected into linear form are then distributed seem-
ingly chaotically and randomly. We dub this hypoth-
esis the ‘Contact Point Hypothesis’.
In order to study this hypothesis, we make one fur-
ther ansatz: namely that the repair process happens
whilst the chromosomes are in their normal spatial ar-
rangement within the cell (i.e. interphase), rather than
during a portion of their lifetime where the chromo-
somes are dramatically repackging themselves. Un-
der this approximation, the spatial mapping is the
same as that extracted from standard HiC techniques
(Lieberman-Aiden et al., 2009).
HiC is a form of Chromatin Conformation Cap-
ture, in which the chromatin strands are crosslinked
with their spatial neighbours, labelled with biotin,
and then excised - producing engineered ’chimeric
reads’, with the chimerism happening preferentially
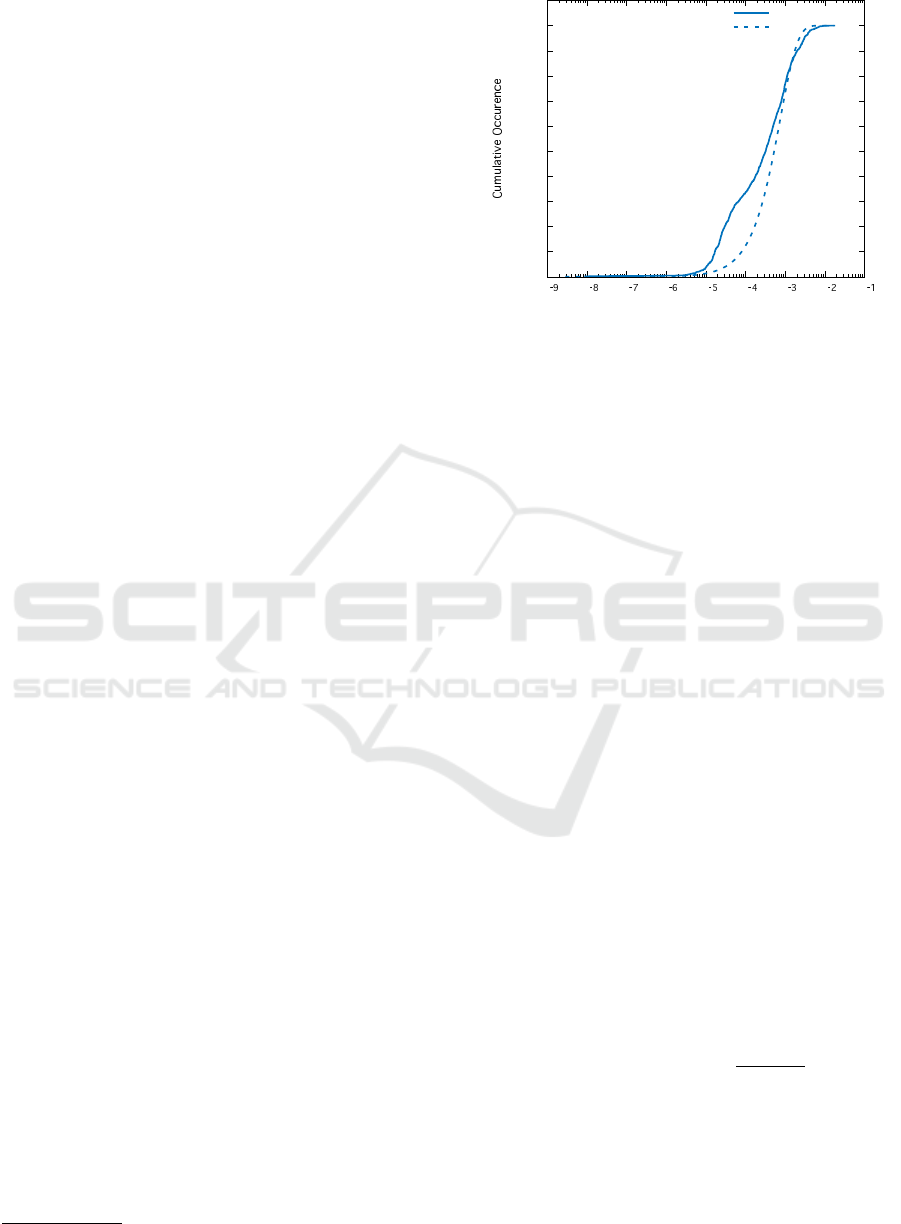
Figure 5: Frequency plots for the values of H
i j
(the contact
number) found at the locations of joinpoints (green), com-
pared to the average value of H
i j
along the corresponding
horizontal of the contact matrix (red).
between reads which are spatially colocated within
the genome. By counting the number of chimerisms
between two regions, one can then build up a HiC
’contact matrix’, H
i j
, which measures how close the
genetic coordinates i and j are in 3D space.
Our hypothesis is equivalent to approximating
that:
Prob(i joins to j) =
H
i j
∑
k
H
i,k
(3)
I.e., the probability of seeing a join at a location is
directly proportional to the HiC contact mapping be-
tween the original ostracon location, and its final po-
sition.
Figure 5 demonstrates the distributions of H
i j
ex-
tracted for the joinpoints in our sample, as compared
to the mean value,
H
i j
=
1
N
∑
N
j
H
i, j
. It seems clear
from this plot that H
i j
<
H
i j
almost everywhere,
and hence that the joins are actually occuring very far
away from regions of high contact. It might also be
tempting to take this one step further, and say that the
breakpoints are preferentially happening away from
regions of high contact.
5.1 Additional Complexity: The Need
For A Full Analysis
However, we note that this is a relatively simple anal-
ysis and omits a potentially vitally important corol-
lary: that we potentially do not observe all joinpoints,
and under the Contact Hypothesis, we would actually
expect to not observe the vast majority of joinpoints.
This is because the ContactPoint hypothesis makes no
distinction between a joinpoint and a perfect repair.
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
280

If the break was repaired perfectly, we would
have no way to detect that it exists, since we can
only detect joinpoints that result in chimeric align-
ment. Such a break would not be counted by the
green line in Figure 5, despite potentially having
contact counts in the thousands. In short, by the
very nature of the observations, we preferentially
omit our most probable datapoints. The probabil-
ity we need to test is not Prob(join at (i, j)), but
Prob(join at (i, j )||i − j| > X), i.e. the breakpoints
must be far enough apart for them to be distinct and
detectable. There are other additional considerations
to take into account: joins within highly repetitive re-
gions are unlikley to be detected due to the difficulty
of accurately aligning to them, for example.
We therefore urge caution in interpreting the raw
data in this fashion, and instead leverage the pow-
erful Bayesian machinery developed in section 3 to
test a number of alternative hypotheses. As noted in
§3, our Bayesian approach is not strictly about ac-
cepting/rejecting a null hypothesis, but about learning
which of a series of proposed models is the best at
describing the data - though we do include a highly
simple model as a pseudo-null, as a baseline against
which all other models are compared.
To this end, we propose three classes of model to
test, corresponding to three basic Hypotheses
1. Hypothesis: There is no pattern: Our null model
(as far as we have one) is the ‘Uniform Weight-
ing’ (UW) model, which assumes that there is
no underlying pattern in the location of the join-
points, and every join is as likely as the others:
p
i j
= C (4)
This model has no free parameters, as the value of
C is determined by the size of the chromosome.
2. Hypothesis: There is a pattern (but we don’t know
what): The next most simple model assumes that
the chromosome can be split up into N segments:
each segment has a uniform probability of a join
occuring within it, but this varies from segment
to segment: a Multi-Block Uniform Weighting
(MBUW) model.
p
i j
= A
xy
= A
yx
(
i in block x
j in block y
(5)
This model has N(N + 1)/2 − 1 free parameters,
corresponding to the number of possible A
i j
val-
ues, minus one for the normalisation. We denote
the models with different resolutions as MBUW
x
3. Hypothesis: The Contact Point Hypothesis is true:
In this case, we use the HiC contact map to gen-
erate a Spatially Associated Weighting (SAW)
model. Since HiC maps are (by nature) sparse,
we pass a Gaussian smoothing kernel of length ℓ
over the map in order to populate all values of p
i j
:
p
i j
= smooth
H
i j
∑
k
H
i,k
,ℓ
(6)
We could equally bin the HiC data into coarser
bins, but for the purposes of marginalisation, it
is often more convenient to deal with a continu-
ous parameter. We label the model which has a
smoothing length of 10
x
bases as SAW
x
.
From each of these proposed models for p
i j
, we
are then able to generate a value of P(D|model), the
probability of observing each chromothripsis dataset
(which we recall is a list of join-points (i
k
, j
k
) of each
ostracon in detected by STEPPINGSTONE), and hence
compute Eq.(1).
Before testing these models, however, it is use-
ful to first discuss what each model being “the best”
would mean. In the case of the SAW model scoring
highly for some reasonable value of ℓ, the conclusion
would be that the Contact Point hypothesis is indeed
a reasonable model for how ostracons are reassem-
beld during chromothripsis. If the UW model scores
highly, it means that all of our proposed models are
less likely than sheer random chance: in this case,
we would probably argue that it is more likely that
we failed to properly formulate a model than the UW
model being ”true” in any meaningful way.
The MBUW models are perhaps the most diffi-
cult to interpret; the most obvious point is that if
a MBUW
x
model is found to outperform both the
UW and SAW models, this implies that there is in-
deed a pattern in the underlying distribution of join-
points, but that it is not the Contact Point hypothe-
sis. However, we can also infer some more informa-
tion, since the MBUW models allow for fine struc-
ture in the probability distribution of the chromosome,
but the so-called ’Occam Factor’ implicit in Eq.(1)
means that arbitrarily high dimensional formulations
are punished. Therefore, if MBUW
x
is found to be a
good fit, but MBUW
x+1
is not, this implies that the
smallest scale of variation in the underlying pattern
is one-x
th
of the size of the chromosome. Testing
the MBUW models of arbitrarily high dimension can
therefore be used to infer the variation scale (but can
be computationally very costly due to the multidimen-
sional integrals required: we limit ourselves to x = 25
- a 324 dimensional integral).
Figure 6 shows the results of such an inference on
three classes of model: For each model, we computed
the integral shown in Eq.(1), relative to the best per-
forming model. Note that for ease of interpretation,
we have inverted the scale: a high value means that
the model has performed poorly.
Ongoing Work to Study the Underlying Statistical Patterns of Oesophageal Chromothripsis
281
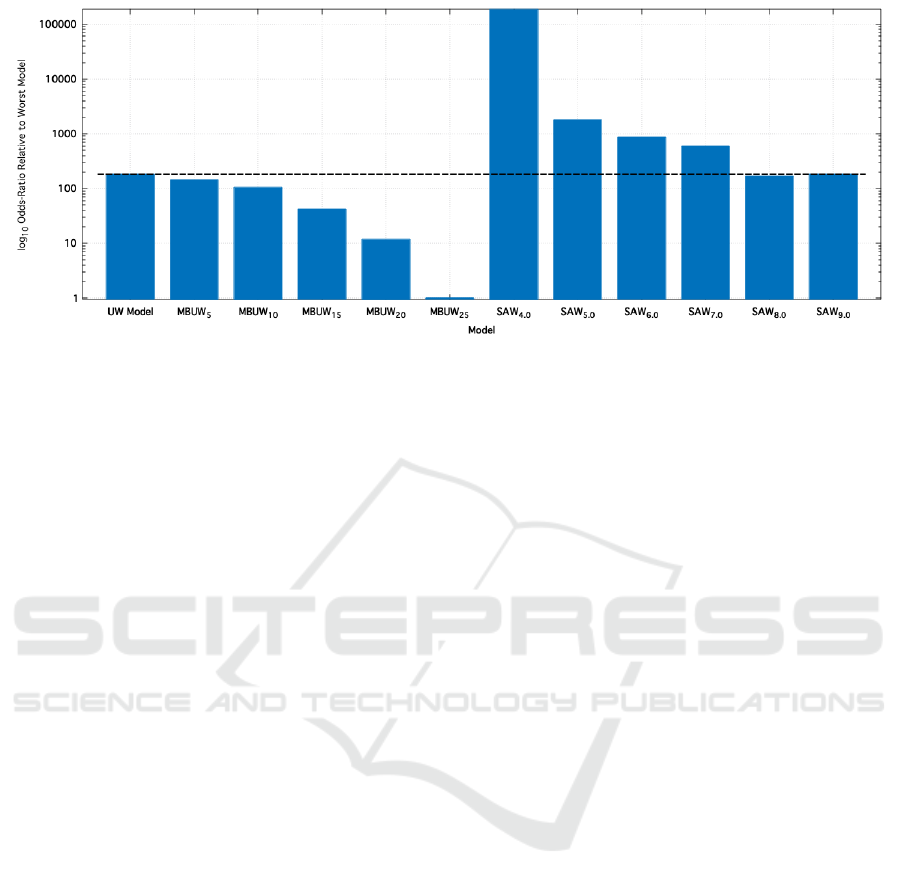
Figure 6: Bayesian inference plot for a number of joinpoint Hypotheses tested against the joinpoint data on chromosome
6 of the OESO-103 dataset. Shown are the values of Eq.(1) computed for each model, normalised such that a value of
x corresponds to a hypothesis 10
x−1
less likely than the most likely one. This demonstrates that even with the additional
considerations detailed in 5.1, the Contact Point hypothesis (detailed by the ‘SAW’ models) provides a significantly worse fit
for the data than assuming no underlying pattern at all (the UW model).
Figure 6 clearly shows that the MBUW models
outperform the UW model, which in turn outperforms
almost all the SAW models. It is only by setting the
blurring distance extremely high (non-trivial portions
of the entire chromosome) that the Contact Point hy-
pothesis even approaches the random-pattern of the
UW model.
We therefore conclude that, given this sample
data, the contact point hypothesis in the form pre-
sented is extremely unlikely to be true. However,
there is significant indication of underlying patterns
within the position of joinpoints: there is variation in
the probability distribution below the order of 6Mb -
a value determined solely by the computational con-
straints of the 234 dimensional integral.
5.2 The End of Contact Points?
This does not necessarily rule out the notion that join-
points are formed from spatial proximity: it merely
rules out that the spatial mapping is the same as that
measured by HiC data. If the process of chromothrip-
sis occurs during a different phase of chromosome ar-
rangement, then the associated spatial mapping would
also be different: this could be a naturally occuring
rearrangement (i.e. anaphase or apoptosis), or due to
some induced change associated only with the chro-
mothripsis mechanism.
To this end, we are also working on a method to
detect Contact Point association without the need for
the pre-generated map, H
i j
. Under this approach, we
merely have to posit that such a matrix exists, and
then marginalise over all possible mappings, with the
dataset expanded to include multiple chromothripsis
samples. If chromothripsis occurs due to a consistent
spatial mapping, we would therefore find a consistent
contact point weighting between the samples.
Of course, in doing so we have no guarantee that
the mapping matrix corresponds to physical proxim-
ity: this would simply demonstrate that there exists a
fixed, underlying mapping between joinpoints across
multiple different instances of chromothripsis, which
is itself an interesting notion.
However, our primary limitation at this time is
a paucity of high quality chromothripsis samples.
Therefore, whilst the statistical machinery is within
reach, we must wait for a larger set of biological sam-
ples.
6 CONCLUSIONS
In this position paper we have detailed a number of
tools and avenues of study that we are developing in
our effort to understand the underlyng statistical prop-
erties of the chromothripsis phenomenon. Although
this is a work in progress and our results only prelim-
inary, we have made great strides in improving our
understanding.
Our HYPOTHESISTESTER tool, though developed
specifically for this work, has the potential to make
Bayesian statistical inference an easy-to-use and ac-
cessible tool in many diverse and distinct fields, and
therefore represents a concrete step towards resolv-
ing a particularly strong tension between embattled
camps in the field of statistical inference.
We have demonstrated how this tool can be lever-
aged to distinguish between different biological mod-
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
282

els, in particular, in the case of the Contact-Point hy-
pothesis, we were able to demonstrate that although
our hypothesis was significantly worse than posit-
ing no structure at all, the statistical mechanism un-
derlying HYPOTHESISTESTER clearly indicated that
there is additional structure present, below the scale
of 6Mb: we are able to confirm that there is a statisti-
cal mechanism to be discovered - we are just not quite
sure what it is yet.
In addition, our work on the length of the ostra-
cons generated by the chromothripsis event provided
a glimpse that, although breakpoints undoubtedly ex-
hibit clustering around a series of nexuses, the distri-
bution of these focal points seems to be random and
uniform across chromosome 6, in strong tension with
some claims we have highlighted from the previous
literature. The presence of a uniform distribution of
focal points seems to lie in contradiction to the oth-
erwise highly structured suprachromosomal pattern
(i.e. chromothripsis on only a single copy of a few
chromosomes), and the clustering around these focal
points, a tension which might help inform future stud-
ies into the actual mechanisms of chromothripsis.
We emphasise again that these are some prelim-
inary results, and aknowledge that we must expand
our data beyond the single, unusually prolific case
of chromothripsis we have studied here. We have
also demonstrated several further steps that need to
be taken from a theoretical perspective, in formualt-
ing more robust and powerful statistical models for
both the ostracon size analysis, and the mapless Con-
tact Point testing. However, despite their preliminary
nature, the results here undoubtedly represent an in-
truiging insight into the future work ahead.
ACKNOWLEDGEMENTS
We want to thank Dr Peter Campbell and Dr Jannat
Ijaz, Wellcome Sanger Institute, for providing the oe-
sophageal cancer datasets in the analysis.
REFERENCES
Fraser-Govil, J. (2023). Hypothesis tester: A flexible tool
for bayesian model inference. Unpublished work.
Fraser-Govil, J. and Boubert, D. (2023). An efficient al-
gorithm for stohastic gradient descent on very large
datasets. Unpublished work.
Korbel, J. O. and Campbell, P. J. (2013). Criteria for in-
ference of chromothripsis in cancer genomes. Cell,
152(6):1226–1236.
Lieberman-Aiden, E., Van Berkum, N. L., Williams, L.,
Imakaev, M., Ragoczy, T., Telling, A., Amit, I., La-
joie, B. R., Sabo, P. J., Dorschner, M. O., et al. (2009).
Comprehensive mapping of long-range interactions
reveals folding principles of the human genome. sci-
ence, 326(5950):289–293.
Maher, C. and Wilson, R. (2012). Chromothripsis and hu-
man disease: Piecing together the shattering process.
Cell, 148(1):29–32.
Mardin, B. R., Drainas, A. P., Waszak, S. M., Weis-
chenfeldt, J., Isokane, M., St
¨
utz, A. M., Raeder, B.,
Efthymiopoulos, T., Buccitelli, C., Segura-Wang, M.,
Northcott, P., Pfister, S. M., Lichter, P., Ellenberg, J.,
and Korbel, J. O. (2015). A cell-based model sys-
tem links chromothripsis with hyperploidy. Molecular
Systems Biology, 11(9):828.
Rausch, T., Jones, D., Zapatka, M., St
¨
utz, A., Zichner, T.,
Weischenfeldt, J., J
¨
ager, N., Remke, M., Shih, D.,
Northcott, P., Pfaff, E., Tica, J., Wang, Q., Massimi,
L., Witt, H., Bender, S., Pleier, S., Cin, H., Hawkins,
C., Beck, C., von Deimling, A., Hans, V., Brors, B.,
Eils, R., Scheurlen, W., Blake, J., Benes, V., Kulozik,
A., Witt, O., Martin, D., Zhang, C., Porat, R., Merino,
D. M., Wasserman, J., Jabado, N., Fontebasso, A.,
Bullinger, L., R
¨
ucker, F. G., D
¨
ohner, K., D
¨
ohner,
H., Koster, J., Molenaar, J., Versteeg, R., Kool, M.,
Tabori, U., Malkin, D., Korshunov, A., Taylor, M.,
Lichter, P., Pfister, S., and Korbel, J. (2012). Genome
sequencing of pediatric medulloblastoma links catas-
trophic dna rearrangements with tp53 mutations. Cell,
148(1):59–71.
Righolt, C. and Mai, S. (2012). Shattered and stitched
chromosomes—chromothripsis and chromoanasyn-
thesis—manifestations of a new chromosome crisis?
Genes, Chromosomes and Cancer, 51(11):975–981.
Solorzano, C. O. S., Pascual-Montano, A., de Diego, A. S.,
Mart
´
ınez-A, C., and van Wely, K. H. (2013). Chro-
mothripsis: Breakage-fusion-bridge over and over
again. Cell Cycle, 12(13):2016–2023. PMID:
23759584.
Stang, A., Poole, C., and Kuss, O. (2010). The ongoing
tyranny of statistical significance testing in biomed-
ical research. European journal of epidemiology,
25(4):225–230.
Stephens, P. J., Greenman, C. D., Fu, B., Yang, F., Bignell,
G. R., Mudie, L. J., Pleasance, E. D., Lau, K. W.,
Beare, D., Stebbings, L. A., McLaren, S., Lin, M.-
L., McBride, D. J., Varela, I., Nik-Zainal, S., Leroy,
C., Jia, M., Menzies, A., Butler, A. P., Teague, J. W.,
Quail, M. A., Burton, J., Swerdlow, H., Carter, N. P.,
Morsberger, L. A., Iacobuzio-Donahue, C., Follows,
G. A., Green, A. R., Flanagan, A. M., Stratton, M. R.,
Futreal, P. A., and Campbell, P. J. (2011). Massive ge-
nomic rearrangement acquired in a single catastrophic
event during cancer development. Cell, 144(1):27–40.
Voronina, N., Wong, J. K., H
¨
ubschmann, D., Hlevnjak, M.,
Uhrig, S., Heilig, C. E., Horak, P., Kreutzfeldt, S.,
Mock, A., Stenzinger, A., et al. (2020). The landscape
of chromothripsis across adult cancer types. Nature
communications, 11(1):1–13.
Ongoing Work to Study the Underlying Statistical Patterns of Oesophageal Chromothripsis
283
