
Formal Analysis of Rewriting System Representing RNA Folding
Krishnendu Ghosh
1 a
and Julia Goldman
2 b
1
Department of Computer Science, College of Charleston, SC, U.S.A.
2
Department of Mathematics, Texas Christian University, TX, U.S.A.
Keywords:
RNA Folding, Probabilistic Model Checking, Rewriting System, Stochastic Modeling.
Abstract:
Prediction of RNA structure is an important problem in understanding biological processes in living organism.
Computational models have been created to study the processes with the aim of unravelling the RNA structure.
In this work, a novel formalism for formal analysis of RNA structure prediction is described. A graph rewrit-
ing system is formalized to represent structural dynamics of RNA structure under uncertainty. Probabilistic
model checking is performed on queries seeking structural properties in RNA. Experiments were conducted
to evaluate the computational feasibility of the model.
1 INTRODUCTION
The significance of the role of RNA in biological pro-
cesses such as gene expression and inhibition is im-
mense (Riddihough, 2016). The structural dynam-
ics of RNA provides insights in the biological pro-
cesses. RNA secondary structure prediction is critical
in understanding the function of RNA. The primary
structure of RNA is represented by a sequence of the
nucleotides- A, U, G, C. The RNA secondary struc-
ture is formed with the folding of an RNA strand with
formation of hydrogen bonds. RNA pseudoknots are
formed from the Watson-Crick base pairing. It is ac-
cepted that the secondary RNA structure is predicted
based on the minimum free energy for stability.
The problem of predicting RNA secondary structure
containing pseudoknots is NP-complete for a large
number class of pseudoknots (Lyngsø and Peder-
sen, 2000). The design of secondary structure us-
ing the Watson-Crick is NP-complete in a more re-
alistic model of RNA sequence (Bonnet et al., 2020).
The prediction of RNA secondary structure is com-
putational intensive and hence, construction of novel
methods are necessitated. Machine learning algo-
rithms have been studied for RNA secondary structure
prediction (Zhao et al., 2021). Given the black-box
nature of deep learning (Sato et al., 2021), it is not that
useful for biologist to understand the complete pro-
cess of the structural dynamics of RNA. Probabilis-
a
https://orcid.org/0000-0002-8471-6537
b
https://orcid.org/0000-0003-1963-5914
tic models have been useful in modeling RNA sec-
ondary structure when different sources of data, such
as homologous RNA sequences, thermodynamic pa-
rameters of the energy minimization model, are com-
bined to predict structure (Dowell and Eddy, 2004).
The goal of this work is to evaluate how the struc-
tural change a RNA will go through during change
of free energy. It is not possible to get precise value
of free energy through experiments for studying the
structural dynamics of RNA. Computational methods
have been sought to study changes in the RNA struc-
ture. Our work leverages on construction of a for-
malism that is based on rewriting system under un-
certainty. The computational challenge is to relate the
structural changes with the minimum free energy. We
consider the minimum free energy as the reason for
the RNA structural changes. The rewriting rules rep-
resents the change in the structure from one structure
to another. The contribution of this work is to model
RNA structural dynamics with a finite state machine
under uncertainty and then, apply temporal logic as a
querying mechanism to evaluate RNA structural dy-
namics.
Model checking is a technique that verifies dynamic
properties on a finite state machine representing the
system. Correctness of software, network protocol
and hardware have been verified using model check-
ing. Model checking represents a system symbol-
ically and not explicitly. The time complexity of
model checking is polynomial to the size of the
model. Properties or specifications are stated in the
form of temporal logic formulas which are precise
Ghosh, K. and Goldman, J.
Formal Analysis of Rewriting System Representing RNA Folding.
DOI: 10.5220/0011734300003414
In Proceedings of the 16th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2023) - Volume 3: BIOINFORMATICS, pages 235-242
ISBN: 978-989-758-631-6; ISSN: 2184-4305
Copyright
c
2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)
235

properties posed as a query to the finite state machine
representation of the system. Probabilistic model
checking is performed which is the properties repre-
sented by different computational logics are posed as
query to the stochastic structures.
We create a formalism to study RNA structural dy-
namics by representing RNA structures by graph
rewriting, and uncertainty in the dynamics is incorpo-
rated by using stochastic models. Stochastic models
represent uncertainty in the model when RNA strands
transitions from one structure to another with dif-
ferent, random rates. Computational feasibility and
properties of the model are evaluated by experimen-
tation using software, PRISM (Kwiatkowska, 2003).
To the best of our knowledge, this is the first work that
demonstrates application of model checking to RNA
structure prediction.
2 RELATED WORK
In this section, we describe the related work on mod-
eling of RNA secondary structure prediction based on
discrete structures and inferences using logic-based
approaches.
Formal language approaches have been investigated
in the modeling of RNA structure. An algebraic lan-
guage for tree representation of RNA secondary struc-
ture was described (Quadrini et al., 2019). The op-
erations defined on the language were concatenation,
nesting, and crossing. Concatenation was used for
motifs where one structure follows another. Nesting
was used to show when a structure had been inserted
into the hairpin, and crossing was to show the interac-
tion between structures. The three operations are used
to create a unique tree representation for each RNA
structure. To ensure that all RNA secondary structures
could be expressed, the operators were used to repre-
sent pseudoknots as a unique combination of hairpins,
the most basic loop structure. A novel method to com-
pare RNA secondary structures using specific rep-
resentations of secondary structures based algebraic
tree has been reported (Quadrini et al., 2020). RNA
pseudoknots have been modeled using term rewriting
(Fu et al., 2008).
Formal grammars have been proposed (Jonoska et al.,
2021) for modeling RNA:DNA interactions and the
formation of R-loops (3-stranded nucleic acid hybrid
structure). RNA folding was modeled as graph trans-
formation in the presence of free energy (Mamuye
et al., 2016). In this model, each RNA configura-
tion was represented as a graph and the evolution of
configurations was rule based, represented by graph
grammar.
SAT solvers were also applied in RNA secondary
structure prediction (Ganesh et al., 2012). The user-
provided code included structural constraints (biolog-
ical properties of the RNA structure) and energy con-
straints (quantitative requirements). Specifically, the
work address correct attribution of a structural state to
each nucleic acid within an RNA sequence. Danos et
al (Danos et al., 2012) construct pathways using a new
graph-based semantics system and a rule-based lan-
guage for protein-protein interactions called Kappa.
Single pushout (SPO) is the technique used for this
model. This means that there will be a left-hand
side, a right-hand side, and a domain of definition.
RNA can be described using an alphabet of the nu-
cleotides, (A,U,G,C) and its secondary structure can
be described by the ways in which the nucleotides
bond with each other. Often, the optimal secondary
structure is predicted to be the one with minimum free
energy (MFE). In the Watson-Crick model this would
be the structure with the most base pairs. The predic-
tion of the RNA structure with MFE is evaluated for
models that do not contain pseudoknots. This is called
the RNA folding problem. Inclusion of pseudoknots
in the problem essentially causes it to be NP-complete
(Bonnet et al., 2020). The RNA design problem in-
volves finding a sequence of nucleotide that folds into
a given secondary structure. RNA Design Extension
is the same, except for the added condition that some
indices of the sequence must contain a specified base.
A sample of Boltzman distribution to generate subop-
timal RNA structures has been reported (Rogers et al.,
2017). Algorithmic construction of RNA secondary
structures was investigated and the result- designing
RNA secondary structures in the Watson-Crick model
was proved to be NP hard if the input structure was la-
beled with bases at some designated position (Bonnet
et al., 2020).
There is a body of literature of model checking in
systems biology, in particular using stochastic mod-
els which has has been an active research area for
a decade (Kwiatkowska and Thachuk, 2014). For-
mal modeling such as model checking has been used
as a querying mechanisms on models of biochemical
pathways (Heath et al., 2008; Chabrier-Rivier et al.,
2004).
3 PRELIMINARIES
In this section, we give the definitions on which the
formalism for RNA structure prediction is based. The
formalism integrates concepts from multiple topics
such as stochastic structures- discrete-time Markov
chain, continuous-time Markov chain, probabilistic
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
236

model checking and graph rewriting.
The state based definition of the stochastic structures
such as discrete time Markov chain (Baier et al.,
2008) is:
Definition 1. (Discrete-Time Markov Chain
(DTMC)) a discrete-time Markov chain is a tu-
ple: M
m
hS,S
0
,ι
init
,P,Li where:
1. S is a finite set of states.
2. S
0
is the set of initial states.
3. P : S × S → [0,1] , where P represents the proba-
bility matrix and
∑
s,s
0
∈S
P (s,s
0
) = 1.
4. ι
init
: S → [0, 1] where
∑
s∈S
ι
init
(s) = 1 is the initial
distribution.
5. L : S → 2
AP
, where L is a labeling function and AP
the set of atomic propositions.
Definition 2. (Labeled Continuous-Time Markov
Chain (LCTMC)) A labeled Continuous-time Markov
Chain (Baier et al., 2008) is a tuple, K =
hS,S
0
,R,AP, Li where:
1. S is a set of states.
2. S
0
⊂ S is the set of initial states.
3. R : S × S → R
≥0
as the rate matrix.
4. L : S ← 2
AP
is a labeling function.
The labeled CTMC described in Definition 2 elim-
inates the requirement R(s,s) =
∑
s6=s
0
R(s,s
0
), unlike
non-state based definition of CTMCs. Self-loops are
modeled by R(s,s
0
) > 0.
Definition 3. (Probabilistic Model checking) Given a
probabilistic model,M
p
and formula,φ ,model check-
ing is the process of computing the answer to the
question of whether M
p
|= φ holds.
PCTL syntax includes state formulas φ and path
formulas ψ. Within the formulas, the next, bounded
until, and until operators are allowed (Parker, 2003).
3.1 Probabilistic Computation Tree
Logic
We describe the syntax and semantics of probabilis-
tic computation tree logic (PCTL) ((Aziz et al., 1995;
Hansson and Jonsson, 1994)).The syntax of PCTL is:
φ ::= true | p | φ ∧ φ | ¬φ | P
⊕J
[ψ]
ψ ::= X φ | φU
≤k
φ | φUφ
where p is an atomic proposition,⊕ ∈ {≤, <,≥, >
},J ∈ [0,1] and k ∈ N. φ,ψ are state and path for-
mula respectively. φ and ψ are state and path for-
mulas respectively. Each of these formulas are inter-
preted over a DTMC or an MDP. Each state of DTMC
or MDP is labeled from the set of atomic proposi-
tion. Specification is represented in the form of a state
formula. Path formula ψ are preceded by the prob-
ability path operator P . Examples of intervals that
are bounds for P are : P
≤0.5
(ψ) denotes P
[0,0.5]
(ψ).
DTMC satisfies P
⊕J
is the probability of a path from
s satisfying ψ is in the bound stated by ⊕p. The path
forumla,Xφ is true if φ is satisfied in the next state.
The formula φ
1
U
≤k
φ
2
is true if φ
2
is satisfied within
k time-steps and φ
1
is true at that point. Similar is the
description of φ
1
Uφ
2
where φ
2
is true some point in
future till then φ
1
is true.
The semantics of PCTL over DTMC is given by:
Given a DTMC, M
p
= hS
0
,S, P ,Li and a PCTL for-
mula, the notation s |= φ represents φ is satisfied in s.
For a given path, π satisfyinng a PCTL path formula,
the notation is π |= ψ. The semantics of PCTL over
M
p
(Parker, 2003):
For a path π :
1. π |= Xφ iff π(1) |= φ.
2. π |= φ
1
U
≤k
φ
2
iff ∃i ≤ k.(π(i) |= φ
2
∧ π( j) |=
φ
1
,∀ j < i.
3. π |= φ
1
Uφ
2
iff ∃k ≥ 0, π |= φ
1
U
≤k
φ
2
For a state, s ∈ S:
1. s |= true,∀s ∈ S.
2. s |= a iff a ∈ L(s).
3. s |= φ
1
∧ φ
2
iff s |= φ
1
∧ s |= φ
2
.
4. s |= ¬φ iff s 6|= φ.
5. s |= P
⊕J
[ψ] iff p
s
(ψ) ⊕ p.
where p
s
(ψ) = Pr
s
({π ∈ Path(s) | π ||= ψ}) where Pr
s
is the set of paths consistes of non-empty sequence of
states in the DTMC.
CTMCs can be described by two properties: tran-
sient behavior and steady-state behavior. Transient
behavior describes the system at a particular moment
in time, whereas steady-state behavior describes the
system in the long-run.
The temporal logic used to specify properties of
CTMCs is called continuous stochastic logic (CSL).
In addition to the operators used in PCTL, CSL also
uses the time-bounded until operator and the steady-
state operator S (Parker, 2003).
3.2 Continuous Stochastic Logic
Model checking on CTMC is performed by continu-
ous stochastic logic (CSL) (Aziz et al., 1996; Baier
et al., 1999). The syntax of CSL (Aziz et al., 1996)
The syntax of CSL is
φ ::= true | a | φ ∧ φ | ¬φ | P
⊕p
[ψ] | S
⊕p
[φ]
ψ ::= X φ | φU
<=k
φ |φUφ
Formal Analysis of Rewriting System Representing RNA Folding
237

where a is an atomic proposition,⊕ ∈ {≤,<,≥, >
}, p ∈ [0,1] and k ∈ R
≥0
. φ,ψ are state and path for-
mula respectively. φ and ψ are state and path formu-
las respectively. P
⊕
pψ]represents the probability of
φ satisfied from a given state satisfies the bound ⊕p.
The bounded until operator φ
1
U
≤
kφ
2
is valid if φ
2
for
a time instant in the interval [0, k] and φ
1
is valid at
all preceding time instants. The other until operator,
U is not dependent. DTMC or MDP satisfies P
⊕p
is
the probability of a path from s satisfying ψ is in the
bound stated by ⊕p .The path formula,Xφ is true if
φ is satisfied in the next state. The formula φ
1
U
≤k
φ
2
is true if φ
2
is satisfied within k time-steps and φ
1
is
true at that point. Similar is the description of φ
1
Uφ
2
where φ
2
is true some point in future till then φ
1
is
true.
3.3 RNA Probabilistic Rewriting
System
We define the language on RNA graphs and graph
rewriting: (G Taentzer and K Ehrig, 2006). The dy-
namics of RNA structure is modeled using rewrit-
ing system. In our formalization, we construct RNA
structural graph and then, create a model that lever-
ages on the graph rules under uncertainty. Our model
integrates representation of RNA graph and uncer-
tainty in the folding of RNA.
Definition 4. An RNA-graph is a graph
G
r
(V,E,L,L
e
) where vertices represent bases ,
and edges represent the bonds between bases such
that
1. V is the set of vertices.
2. E is the set of edges and e ∈ E. e = hv, v
0
i and
v,v
0
∈ V .
3. (No self loop) 6 ∃hv,ui such that v = u.
4. (Labeling function) L : V → Ba where Ba is set of
bases and Ba={A,G,U,C}.
5. (Edge Labeling function) L
e
: V → Bo where Bo is
the set of bonds.
In the construction of the graph rewriting system,
the set of rules are triggered by a probability (Krause
and Giese, 2012). The probabilistic folding model
is adapted from probabilistic timed graph (Maximova
et al., 2018) where Dist(Z) is the set of probability
distribution on the set of rules, Z.
Definition 5. (Probabilistic folding rule) A proba-
bilistic folding rule, κ = hG,Z,µi where
1. G is the RNA graph.
2. Z is the set of non-empty finite rules such that G =
L for all z = hL
l
← K
r
→ Ri ∈ Z, and µ ∈ Dist(Z).
Here, there are multiple right-hand sides,R for
a single G. For the RNA structure model, Z =
{hairpin, bulge, helix, internal loop}.
Rewrite rules generate graph transformations.
Our model focuses on representing the secondary
structure of RNA molecules. Secondary structure
refers to the ordered sequence of bases and the bonds
that connect them.
3.4 Model
The model based on discrete-time Markov chain,M is
the following- A RNA graph, call it a starting graph,
G
0
is transformed
ˆ
G
0
in next state, when one of the
rules, z ∈ Z triggers. The reading of a transition from
a state, s to state s
0
is a RNA graph G
0
under a rule,z
is transformed into
ˆ
G
0i
with probability, p
i
such that
the sum of p
i
is 1. Here, s,s
0
∈ S where S is the set
of states in M . For the CTMC variant, there is no re-
quirement of sum of p
i
s should be one. The rates are
the labels on the transition. The reading of a transition
from a state, s to state s
0
is a RNA graph G
0
under a
rule, z is transformed into
ˆ
G
0i
with rate, q.
The finite state machine,K of the RNA structural dy-
namics is represented as: Given a finite set of RNA
structure,S
rna
= {st
1
,st
2
,. .. st
n
} and set of finite min-
imum free energy, FE = { f e
1
, f e
2
,. .. , f e
m
} where
f e
i
∈ R and n,m, i ∈ N. The states of K are la-
beled with a structure, st, st
0
∈ S
rna
and f e ∈ FE. A
transition, s → s
0
where s,s
0
are states in K implies
structure,st is transformed to st
0
in the presence of f e.
Here, the label of s is st and f e. The label of s
0
is st
0
.
4 SIMULATION
4.1 Data Preparation
The data for the simulation was from RNAeval web-
server (RNA, ) in a model of RNA structure(Mamuye
et al., 2016), The program, part of the ViennaRNA
Web Services(Gruber et al., 2008), allows the user to
input any RNA sequence, and then server calculates
the energy on a given secondary structure. The
thermodynamic description given by RNAeval to
calculate the free energy of each structure (Mamuye
et al., 2016). The free energy values make it possible
to choose the optimal structure for each step in the
graph transformation. The comparison of predicted
optimal structure to the one predicted by the RNAfold
web server were validated (Mamuye et al., 2016).
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
238
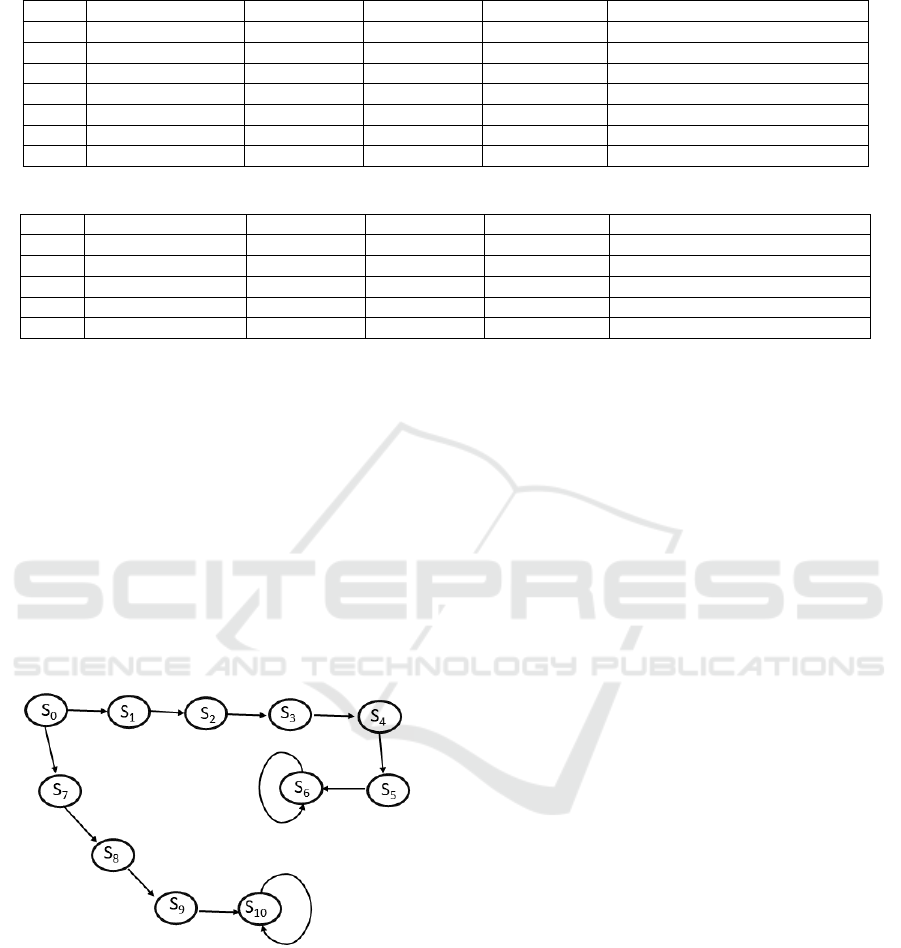
Table 1: Path from state 0 to state 6.
State Energy(kcal/mol) w(i) w(p) E(p) Rate from current to next state
0 0.00 1 1 0 1.80E-4
1 4.80 0.000414653 1.000414653 -0.000110966 3.91E-3
2 2.90 0.009047271 1.009461924 -0.002520743 1.25E-3
3 3.60 0.002905783 1.012367707 -0.00329013 1.89E-6
4 7.60 4.41232E-6 1.012372119 -0.003291296 8.15E-6
5 6.70 1.90043E-5 1.012391124 -0.003296321 3.62E-6
6 7.20 8.44358E-6 1.012399567 -0.003298553 terminal state
Table 2: Path from state 0 to state 10.
State Energy (kcal/mol) w(i) w(p) E(p) Rate from current to next state
0 0.00 1 1 0 6.35E-3
7 2.60 0.014720154 1.014720154 -0.003911389 7.64E-4
8 3.90 0.001785947 1.0165061 -0.004382081 1.19E-1
9 0.70 0.321177882 1.337683982 -0.77875129 1.85
10 -2.80 93.9761137 95.31379768 -1.219807747 terminal state
4.2 Computational Feasibility of the
Model
The sample strand, CUUACCAUCGGGUUAGAG-
GAG, used for both the DTMC and CTMC model
is taken from literature (Mamuye et al., 2016). The
energy values of each structure were calculated using
the RNAeval server (Gruber et al., 2008). Figure 1
outlines two possible paths in a finite state machines
for the RNA folding. The structural dynamics of RNA
strand begins in the unfolded state, s
0
. States s
6
and
s
10
have self loop which implies that there is no fur-
ther folding.
Figure 1: Representation of RNA folding in a finite state
machine.
Each arrow represents the formation of a loop
and the bonding of bases, and each subsequent state
represents the resulting change in the molecule’s
secondary structure. The two paths in the finite state
machines are as follows:
Path from s
0
to s
6
:
1. s
0
→ s
1
: Bonds form between the 1
st
and 21
st
bases and the 2
nd
and 20
th
bases to form a helix.
2. s
1
→ s
2
: A bond forms between the 3
rd
and 19
th
bases to form a helix.
3. s
2
→ s
3
: A bond forms between the 6
th
and 16
th
bases to form an internal loop.
4. s
3
→ s
4
: A bond forms between the 7
th
and 13
th
bases to form a bulge.
5. s
4
→ s
5
: A bond forms between the 8
th
and 12
th
bases to form a hairpin.
6. s
5
→ s
6
: A bond forms between the 5
th
and 18
th
bases to form a bulge.
Path from s
0
to s
10
:
1. s
0
→ s
7
: Bonds form between the 1
st
and 16
th
bases and the 2
nd
and 15
th
bases to form a helix.
2. s
7
→ s
8
: A bond forms between the 4
th
and 13
th
bases to form an internal loop.
3. s
8
→ s
9
: A bond forms between the 5
th
and 12
th
bases to form a helix.
4. s
9
→ s
10
: A bond forms between the 6
th
and 11
th
bases to form a helix.
Each path is constructed by minimum free energy
whose values differ and hence, there are two paths
starting from s
0
. In the simulation, the structures
of RNA are represented symbolically. Note that the
sample strand and paths in the finite state machine
representing RNA structural is a simple example and
the simulations results can be validated by compar-
ing to published values. The sample queries in the
form of logic specifications are posed on the stochas-
tic structures representing RNA structural dynamics.
The structures are denoted by the states in the queries.
The experiments were conducted on system with Intel
Core i7 with CPU 2.11 GHz and 16GB RAM. Table 1
and 2 for calculated values of rates and energy where
Formal Analysis of Rewriting System Representing RNA Folding
239

PCTL Formula Results Time (seconds)
P = ? [F x]
”What is the probability of reaching x?
0.027397281 when x = s
6
0.972602719 when x = s
10
0.001 when x = s
6
0.01 when x = s
1
0
P >0.5 [F s=x]
”Verify that the probability of reaching x is greater than 0.5.”
false when x = s
6
true when x = s
10
0.002 when x = s
6
0.009 when x = s
10
P = ? [s
10
U s
6
]
What is the probability that the s
10
is reached before s
6
?
0.027397281 0.006
P = ? [s
6
U s
10
]
”What is the probability s
6
is reached before state s
10
?”
0.972602719 0.008
P = ? [s
3
U s
2
]
”What is the probability that s
3
before s
2
?”
0.00 0.005
Figure 2: Execution times and results for PCTL queries on the DTMC model.
CSL Formula Results Time (sec)
P = ? [F x
6
]
meaning: ”What is the probability that the molecule will reach x
6
?
0.027397281 0.005
P = ? [F x
10
]
meaning: ”What is the probability that the molecule will reach x
10
?”
0.972602719 0.002
P = ? [true U[4,4] x
6
]
meaning: ”What is the probability x
6
exists at time instant 4?”
2.772E-25 0.003
P = ? [true U[4,4] x
10
]
meaning: ”What is the probability of the x
10
at time instant 4?”
2.948E-6 0.002
Figure 3: Execution times and results for CSL queries on the CTMC model.
E(p), w(i),w(p) denote the energy of the path, weight
of the ith state and weight of the path, respectively.
4.2.1 DTMC Model
In the DTMC model, each structure Each transition is
assigned a probability, defined by the equation given
in (Kirkpatrick et al., 2013).
Definition 6. The equilibrium probability for each
state is defined by
e
−E(i)/RT
∑
j∈S
e
−E( j)/RT
where:
1. S is the set of states.
2. i, j ∈ S
3. E(i) is the energy of state i.
4. R is the gas constant. In this case, R is the prod-
uct of Avogadro’s number and the Boltzmann con-
stant.
5. T is the temperature. For this model, T is approx-
imately the body temperature, 310.15 K.
The DTMC model is used to compute the prob-
ability of the molecule terminating at either s
6
or
s
10
. The probability of the molecule reaching s
6
is
0.027397281. The probability of the molecule reach-
ing s
10
, the minimum free energy (MFE) structure,
is 0.972602719. The model checker can also indi-
cate whether a structure is likely to occur by verify-
ing whether the probability is greater than one half.
This is true only when the final structure is state 10.
Additionally, the model can find the probability that
one path will terminate before the other, i.e. the prob-
ability that s
6
forms before s
10
and vice versa. An
observation that the probability s
6
forms before s
10
is
0.027397281, and the probability that s
10
forms be-
fore s
6
is 0.972602719. Similarly, queries can con-
firm the order of states which represents the order of
structures formed.. For example, the probability that
s
3
forms before s
2
is 0. Figure 2 for execution times
for property verification on the model. The sample
queries are reachability queries. The time of execu-
tion of this simple model is within 1 second.
4.2.2 CTMC Model
In the CTMC model, each structure is represented is
labeled to a state. The rates are used from Figure 1
and Figure 2. The CTMC model incorporates tran-
sition rates that were calculated by following the pro-
cess outlined in previous work (Entzian and Raden,
2020): The steps to calculate the transition rates are:
1. The Boltzmann weight, w(i), of each structure
is calculated. The weight is defined by w(i) =
e
−E(i)/RT
. The terms i, E(i), R, and T are as pre-
viously stated in Definition 6.
2. The weight of the path, w(p) is calculated and is
the sum of the Boltzmann weights of all structures
up to and including that point in the path.
3. The energy of the path is calculated and is defined
by E(p) = −RT log(w(p)).
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
240

4. The transition rate is defined by a Metropolis rate,
represented by min(1,
E(p)−E(p
0
)
RT
).
In addition to probabilities, the CTMC model can
be used to incorporate time. For instance, at time in-
stant 4, the probability of the RNA molecule exist-
ing in s
6
is 2.772e − 25. Figure 3 shows the times
recorded on sample CSL queries on the simulation
model. The times for execution for the sample queries
is less than 0.01 second. The computational feasi-
bility of the model is efficient for the simple model.
Therefore, experiments can be performed on large
problem sizes.
5 CONCLUSION
The formalism for RNA structure prediction using
graph rewriting provided insights how a computa-
tional feasible model can be implemented. The model
also demonstrates how uncertainty can be incorpo-
rated in the model and can be quantified in terms of
the probabilities. A model defined by rewriting rules
in the PRISM model checker will become more useful
when different initial RNA strands are used as input
for validation for the formalism. The PCTL and CSL
logics are able to express different but complicated
properties of the system. The formalism provides a
foundation for a rigorous evaluation of RNA structure
prediction. Future work would include experiments
on large datasets of RNA structure.
ACKNOWLEDGEMENTS
A part of this project was supported by grant
P20GM103499-20 (SC-INBRE) from the National
Institute of General Medical Sciences, National In-
stitutes of Health (NIH). Its contents are solely the
responsibility of the authors and do not necessarily
represent the official views of the NIH. KG was sup-
ported by NSF CCF-2227898 for part of the work.
REFERENCES
RNAeval Webserver. http://rna.tbi.univie.ac.at/cgi-bin/
RNAWebSuite/RNAeval.cgi , last accessed on
10/17/2022.
Aziz, A., Sanwal, K., Singhal, V., and Brayton, R. (1996).
Verifying continuous time markov chains. In Inter-
national Conference on Computer Aided Verification,
pages 269–276. Springer.
Aziz, A., Singhal, V., Balarin, F., Brayton, R. K., and
Sangiovanni-Vincentelli, A. L. (1995). It usually
works: The temporal logic of stochastic systems. In
International Conference on Computer Aided Verifi-
cation, pages 155–165. Springer.
Baier, C., Katoen, J.-P., and Hermanns, H. (1999). Approx-
imative symbolic model checking of continuous-time
markov chains. In International Conference on Con-
currency Theory, pages 146–161. Springer.
Baier, C., Katoen, J.-P., and Larsen, K. G. (2008). Princi-
ples of model checking. MIT press.
Bonnet, E., Rzazewski, P., and Sikora, F. (2020). Designing
rna secondary structures is hard. Journal of Computa-
tional Biology, 27(3):302–316.
Chabrier-Rivier, N., Chiaverini, M., Danos, V., Fages, F.,
and Sch
¨
achter, V. (2004). Modeling and querying
biomolecular interaction networks. Theoretical Com-
puter Science, 325(1):25–44.
Danos, V., Feret, J., Fontana, W., Harmer, R., Hayman,
J., Krivine, J., Thompson-Walsh, C., and Winskel, G.
(2012). Graphs, rewriting and pathway reconstruc-
tion for rule-based models. In FSTTCS 2012-IARCS
Annual Conference on Foundations of Software Tech-
nology and Theoretical Computer Science, volume 18,
pages 276–288.
Dowell, R. D. and Eddy, S. R. (2004). Evaluation of several
lightweight stochastic context-free grammars for rna
secondary structure prediction. BMC bioinformatics,
5(1):1–14.
Entzian, G. and Raden, M. (2020). pourrna—a time-and
memory-efficient approach for the guided exploration
of rna energy landscapes. Bioinformatics, 36(2):462–
469.
Fu, X., Wang, H., Harrison, R. W., and Harrison, W. L.
(2008). A rule-based approach for rna pseudoknot
prediction. International journal of data mining and
bioinformatics, 2(1):78–93.
G Taentzer, U. P. and K Ehrig, H. E. (2006). Fundamen-
tals of algebraic graph transformation. with 41 figures
(monographs in theoretical computer science. an eatcs
series).
Ganesh, V., O’donnell, C. W., Soos, M., Devadas, S., Ri-
nard, M. C., and Solar-Lezama, A. (2012). Lynx:
A programmatic sat solver for the rna-folding prob-
lem. In International Conference on Theory and
Applications of Satisfiability Testing, pages 143–156.
Springer.
Gruber, A. R., Lorenz, R., Bernhart, S. H., Neub
¨
ock, R.,
and Hofacker, I. L. (2008). The vienna rna websuite.
Nucleic acids research, 36(suppl 2):W70–W74.
Hansson, H. and Jonsson, B. (1994). A logic for reasoning
about time and reliability. Formal aspects of comput-
ing, 6(5):512–535.
Heath, J., Kwiatkowska, M., Norman, G., Parker, D., and
Tymchyshyn, O. (2008). Probabilistic model checking
of complex biological pathways. Theoretical Com-
puter Science, 391(3):239–257.
Jonoska, N., Obatake, N., Poznanovi
´
c, S., Price, C., Riehl,
M., and Vazquez, M. (2021). Modeling rna: Dna hy-
brids with formal grammars. In Using Mathematics
to Understand Biological Complexity, pages 35–54.
Springer.
Formal Analysis of Rewriting System Representing RNA Folding
241

Kirkpatrick, B., Hajiaghayi, M., and Condon, A. (2013).
A new model for approximating rna folding trajecto-
ries and population kinetics. Computational Science
& Discovery, 6(1):014003.
Krause, C. and Giese, H. (2012). Probabilistic graph trans-
formation systems. In International Conference on
Graph Transformation, pages 311–325. Springer.
Kwiatkowska, M. (2003). Model checking for probability
and time: from theory to practice. In 18th Annual
IEEE Symposium of Logic in Computer Science, 2003.
Proceedings., pages 351–360. IEEE.
Kwiatkowska, M. and Thachuk, C. (2014). Probabilis-
tic model checking for biology. In Software Systems
Safety, pages 165–189. IOS Press.
Lyngsø, R. B. and Pedersen, C. N. (2000). Pseudoknots
in rna secondary structures. In Proceedings of the
fourth annual international conference on Computa-
tional molecular biology, pages 201–209.
Mamuye, A., Merelli, E., and Tesei, L. (2016). A graph
grammar for modelling rna folding. Electronic Pro-
ceedings in Theoretical Computer Science, 231:31–
41.
Maximova, M., Giese, H., and Krause, C. (2018). Prob-
abilistic timed graph transformation systems. Jour-
nal of logical and algebraic methods in programming,
101:110–131.
Parker, D. A. (2003). Implementation of symbolic model
checking for probabilistic systems. PhD thesis, Uni-
versity of Birmingham.
Quadrini, M., Tesei, L., and Merelli, E. (2019). An alge-
braic language for rna pseudoknots comparison. BMC
bioinformatics, 20(4):1–18.
Quadrini, M., Tesei, L., and Merelli, E. (2020). Aspralign: a
tool for the alignment of rna secondary structures with
arbitrary pseudoknots. Bioinformatics, 36(11):3578–
3579.
Riddihough, G. (2016). Signals in rna. Science,
352(6292):1406–1407.
Rogers, E., Murrugarra, D., and Heitsch, C. (2017). Con-
ditioning and robustness of rna boltzmann sampling
under thermodynamic parameter perturbations. Bio-
physical journal, 113(2):321–329.
Sato, K., Akiyama, M., and Sakakibara, Y. (2021). Rna sec-
ondary structure prediction using deep learning with
thermodynamic integration. Nature communications,
12(1):1–9.
Zhao, Q., Zhao, Z., Fan, X., Yuan, Z., Mao, Q., and Yao, Y.
(2021). Review of machine learning methods for rna
secondary structure prediction. PLoS computational
biology, 17(8):e1009291.
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
242
