
Patient Similarity Networks Integration for Partial Multimodal Datasets
Jessica Gliozzo
1,2 a
, Alex Patak
2 b
, Antonio Puertas-Gallardo
2 c
, Elena Casiraghi
1 d
and Giorgio Valentini
1,3 e
1
AnacletoLab - Computer Science Department, Universit
´
a degli Studi di Milano, Via Celoria 18, 20135, Milan, Italy
2
European Commission, Joint Research Centre (JRC), Ispra, Italy
3
ELLIS - European Laboratory for Learning and Intelligent Systems, Milan Unit, Via Celoria 18, 20135, Milan, Italy
Keywords:
Patient Similarity Networks, Similarity Network Fusion, Data Integration, Multi-Omics Data Integration,
Missing Data, Partial Samples.
Abstract:
Integration of partial samples in Patients Similarity Networks, i.e. the combination of multiple data sources
when some of them are completely missing in some samples, is a largely overlooked problem in the multi-
omics data integration literature for Precision Medicine. Nevertheless in clinical practice it is quite usual that
one or more types of data are missing for a subset of patients. We present an algorithm able to combine multiple
sources of data in Patients Similarity Networks when data of one or more sources are completely missing for
a subset of patients. The proposed approach relies on a message-passing learning strategy to recover and
combine completely missing data leveraging the Similarity Network Fusion algorithm. Preliminary results on
TCGA breast cancer data show the effectiveness of the proposed approach.
1 INTRODUCTION
In the last decade, Patient Similarity Networks (PSN)
emerged as a convenient model to integrate multiple
data views and perform clustering/classification tasks
to support Precision Medicine (PM) (Pai and Bader,
2018; Gliozzo et al., 2022). The aim of PM is to tailor
the diagnosis, prognosis and treatment of each patient
making decisions based on her/his genomic, environ-
mental and lifestyle data. This approach represents
a paradigm shift from the use of broad disease cate-
gories typical of a “one size fits all” method (Akhoon,
2021). PM requires by definition the use of big het-
erogeneous data acquired from each patient at differ-
ent levels (e.g. clinical, omics, images, etc), which
is currently possible thanks to the advent of high-
throughput technologies (Lightbody et al., 2019).
PSN are a simple yet powerful way to exploit such
diverse array of data sources. A PSN is a graph where
nodes are patients and edges represent the pairwise
a
https://orcid.org/0000-0001-7629-8112
b
https://orcid.org/0000-0002-9282-188X
c
https://orcid.org/0000-0003-3457-4777
d
https://orcid.org/0000-0003-2024-7572
e
https://orcid.org/0000-0002-5694-3919
similarity between individuals computed using their
clinical and/or biomolecular profiles. The underlying
assumption is that patients described by similar pro-
files should show a similar clinical outcome (Gliozzo
et al., 2022). PSN are suitable for heterogeneous data
since they can be computed from every data type (Pai
and Bader, 2018) providing an overview of the re-
lationships among patients across the different data
views.
The integration of the computed PSN is not triv-
ial and many methods were proposed in literature to
tackle this issue, which are collectively classified as
“PSN-fusion methods” (Gliozzo et al., 2022). Sur-
prisingly, the vast majority of methods require “com-
plete datasets” having all data sources for every con-
sidered sample. However, it is quite common to
have completely missing data sources in multi-omics
datasets (Rappoport and Shamir, 2019; Xu et al.,
2021) due to the limited availability of samples, cost
of the assays and experimental design (Conesa and
Beck, 2019). A naive strategy to integrate multi-
modal data having partial samples, i.e. samples with
one or more data sources not available, is to re-
move them from the dataset. While this approach
is simple, it can significantly reduce the amount of
samples available for the integration and for further
228
Gliozzo, J., Patak, A., Puertas-Gallardo, A., Casiraghi, E. and Valentini, G.
Patient Similarity Networks Integration for Partial Multimodal Datasets.
DOI: 10.5220/0011725500003414
In Proceedings of the 16th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2023) - Volume 3: BIOINFORMATICS, pages 228-234
ISBN: 978-989-758-631-6; ISSN: 2184-4305
Copyright
c
2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)

analysis. More sophisticated approaches attempt to
impute missing data exploiting information coming
from other data sources (e.g. KNN imputation on the
concatenated data matrices (Rappoport and Shamir,
2019)). Unfortunately state-of-the-art approaches to
recover missing data usually work when only some
parts of the data are missing (e.g. when only a rela-
tively small subset of the gene expression data for a
given patient are missing), but are not able to recover
a completely missed source of information for a given
patient (Xu et al., 2021).
Another related open problem regards the devel-
opment of methods to integrate “partial datasets”, i.e.
datasets having individuals with completely missing
data sources. Hence performing a good quality im-
putation and partial samples data integration in PSNs
represents an open problem, largely overlooked in
literature (Rappoport and Shamir, 2019; Xu et al.,
2021).
In this work, we propose miss-SNF: a novel al-
gorithm that can integrate partial datasets through the
cross-diffusion of information among PSN computed
from different data sources. Differently from other
proposed approaches able to handle partial datasets,
miss-SNF can partially reconstruct missing data by
using information from different sources during the
cross-diffusion process.
2 METHODS
Miss-SNF is a novel PSN-fusion method able to com-
bine multiple biological data sources having partial
samples. In particular, miss-SNF leverages the Simi-
larity Network Fusion (SNF) algorithm (Wang et al.,
2014) to integrate two or more data sources and man-
ages partial samples using a message-passing learning
strategy to recover and combine completely missed
data.
2.1 SNF
SNF is a method that exploits a cross-diffusion pro-
cess to pass information among PSN built from dif-
ferent data sources, until convergence to an integrated
network. The first step of SNF is the computation
of a PSN W , expressing the pairwise similarity be-
tween the biomolecular profiles of individuals x
i
and
x
j
, from each data modality. A scaled exponential
similarity kernel is exploited to compute similarity for
continuous data:
W (i, j) = exp
−
ρ(x
i
,x
j
)
2
µε
i j
(1)
where ρ(x
i
,x
j
) is the Euclidean distance between
patients, µ is a hyperparameter related to the variance
of the local model and ε
i, j
is a scaling factor taking
into account the neighbourhoods N
i
and N
j
of the con-
sidered patients:
ε
i, j
=
mean(ρ(x
i
,N
i
)) + mean(ρ(x
j
,N
j
)) + ρ(x
i
,x
j
)
3
(2)
From the initial PSN W (i, j), other two matrices
P and S are computed for each data modality. The
“global” similarity matrix P, which is essential to cap-
ture the overall relationships between patients and it
is computed through the following normalization:
P(i, j) =
(
W (i, j)
2
∑
k̸=i
W (i,k)
, if j ̸= i
1/2 ,if j = i
(3)
where for equation 3 the property
∑
j
P(i, j) = 1
holds. Then a “local” similarity matrix is obtained as
follows:
S(i, j) =
(
W (i, j)
∑
k∈N
i
W (i,k)
, if j ∈ N
i
0 ,otherwise
(4)
where N
i
= {x
k
|x
k
∈ kNN(x
i
) ∪ {x
i
}}. S is able
to capture the local structure of the network because
considers only local similarities in the neighbourhood
of each individual, setting to zero all the others.
Given m data modalities, m different W , S and P
matrices are constructed and an iterative process is ap-
plied where similarities are diffused through the Ps
until convergence, that is, until all the matrices P be-
come similar. In the simplest case, when m = 2, we
have P
(v)
t
that refers to P matrices for data v ∈ {1, 2} at
time t. In this case, the following recursive updating
formulas describes the diffusion process:
P
(1)
t+1
= S
(1)
× P
(2)
t
× S
(1)T
P
(2)
t+1
= S
(2)
× P
(1)
t
× S
(2)T
(5)
In other words P
(1)
is updated by using S
(1)
from
the same data source but P
(2)
from a different view
and vice-versa.
SNF can be easily extended to m > 2 data sources:
P
(s)
= S
(s)
∑
k̸=s
P
(k)
m − 1
S
(s)T
(6)
and the final “consensus” matrix P
(c)
is:
P
(c)
=
1
m
m
∑
k=1
P
(k)
(7)
Patient Similarity Networks Integration for Partial Multimodal Datasets
229

2.2 Miss-SNF
As above-mentioned, a common issue in biological
multi-modal datasets regards the presence of partial
samples, i.e. samples that present one or more com-
pletely missing data sources. The original SNF algo-
rithm (described in the previous section 2.1) requires
the presence of all data modalities for each sample
to perform integration. A naive solution to this prob-
lem cannot consist in setting the feature vector of the
missing data source to
¯
0 (vector of zeros). Indeed it
is easy to see that if we use any similarity measure to
compute the weights we obtain a value different from
zero.
To tackle this problem, we propose two different
extensions of SNF:
1. Reconstruction of missing data by propagation of
the information from other available sources. This
approach is able to partially reconstruct missing
data by using information from different sources
during the cross-diffusion process performed by
SNF. This can be accomplished by appropriately
setting the initial values for W
(s)
,P
(s)
and S
(s)
ma-
trices for the patients having no data for the source
s and then by run the SNF algorithm.
2. Managing missing data by ignoring them. This
second solution simply ignores the missing data
by setting to zero all the entries of the patient x
i
in
the matrices W
(s)
, P
(s)
and S
(s)
and then by run-
ning the vanilla SNF.
2.2.1 Miss-SNF with Partial Reconstruction of
Missing Data (miss-SNF ONE)
The first solution, that handles missing data by par-
tially reconstructing them during the diffusion process
of SNF, is performed by changing the similarity ma-
trices W , P and S. If for a patient x
i
we have a com-
pletely missing source s, the similarity matrices are
modified as follows:
• set W
(s)
(i, j) = 0 ∀ j ̸= i and W
(s)
(i,i) = 1
• set P
(s)
(i, j) = 0 ∀ j ̸= i and P
(s)
(i,i) = 1
• set S
(s)
(i, j) = 0 ∀ j ̸= i and S
(s)
(i,i) = 1
Having a second data source s
′
̸= s, this implies
that the update equation for node x
i
will be:
1. when k ̸= i
P
(s)
t+1
(i, j) =
∑
k∈N
i
∑
l∈N
j
S
(s)
(i,k)S
(s)
( j,l)P
(s
′
̸=s)
t
(k, l) = 0
2. when k = i
P
(s)
t+1
(i, j) =
∑
l∈N
j
S
(s)
(i,i)S
(s)
( j,l)P
(s
′
̸=s)
t
(i,l) > 0,
if ∃l s.t. S
(s)
( j,l) > 0 and P
(s
′
̸=s)
t
(i,l) > 0
The result of the second equation can be different
from 0 when there is a common neighbour x
l
between
x
i
and x
j
such that S
(s)
( j,l) > 0 and P
s
′
̸=s
t
(i,l) > 0.
In other words, we have a contribution to the missing
P
(s)
t+1
(i, j) when does exist a common neighbour be-
tween i and j in respectively the “global” network P
for a different source s
′
̸= s and in the “local” network
S for the missed source s.
This implies that we can populate P
(s)
(i, j) also
when data are missed for source s.
This procedure can be easily extended to manage
missing data having m different sources. Indeed, the
update equation can be written as:
P
(s)
t+1
(i, j) =
∑
k∈N
i
∑
l∈N
j
S
(s)
(i,k)
∑
v̸=s
P
(v)
t
(k, l)
m − 1
!
S
(s)
( j,l)
Referring to
∑
v̸=s
P
(v)
t
(k,l)
m−1
as P
v̸=s
t
(k, l), and having
set S
(s)
(i,k) = 0 ∀ k ̸= i and P
(s)
(i,i) = 1, then:
P
(s)
t+1
(i, j) =
∑
k∈N
i
∑
l∈N
j
S
(s)
(i,k) P
v̸=s
t
(k, l)
S
(s)
( j,l) =
=
∑
l∈N
j
S
(s)
(i,i) P
v̸=s
t
(i,l) S
(s)
( j,l) =
=
∑
l∈N
j
P
v̸=s
t
(i,l) S
(s)
( j,l)
According to the above equations, if x
j
is not
missing, then P
(s)
t+1
(i, j) is “imputed” if x
i
and x
j
share
common neighbours respectively in the “global” net-
work P
v̸=s
and in the “local” network S
(s)
.
2.2.2 Miss-SNF Ignoring Partial Samples
(Miss-SNF ZERO)
The second solution, which handles missing data by
ignoring partial samples in the diffusion process, is
again performed by changing the similarity matrices
W , P and S. If for a patient x
i
we have no data for
source s, the similarity matrices are modified as fol-
lows:
• set W
(s)
(i, j) = 0 ∀ j
• set P
(s)
(i, j) = 0 ∀ j
• set S
(s)
(i, j) = 0 ∀ j
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
230

In this way, the following update rule is obtained:
∀ j : P
(s)
t+1
(i, j) =
∑
k∈N
i
∑
l∈N
j
S
(s)
(i,k)S
(s)
( j,l)P
(s
′
̸=s)
t
(k, l)
= 0
Hence, there will be no contribution to P
(s)
t
(i, j)
because it will remain P
(s)
t
(i, j) = 0 for every value of
t.
The final integrated “consensus” P
(c)
is computed
as:
P
(c)
=
m
∑
k=1
P
(k)
!
⊙ M.
where ⊙ is a pointwise multiplication, and M is a
matrix of the same dimension of P where M(i, j) is the
reciprocal of the sources s available for the edge (i, j).
In other words M(i, j) counts how many sources are
available for the edge (i, j). Note that M = M
T
.
In this way, we obtain a “consensus” P
(c)
that av-
erages edge weights with respect to the actually avail-
able data source for each patient.
3 RESULTS
We present some preliminary results to show the
effectiveness of the proposed approach. By using
multi-omics data from The Cancer Genome Atlas
(TCGA) (Hutter and Zenklusen, 2018), we compared
SNF using the complete data sets with miss-SNF
when amputed data are used instead. We both com-
pared predictions on early/late stage cancer patients
and the resulting integrated adjacency matrices to es-
timate the recovery capabilities of miss-SNF when
partial samples are present in the available data.
3.1 Dataset
To evaluate the performance of miss-SNF, a breast
cancer multi-omics dataset from TCGA (Tomczak
et al., 2015) was downloaded through the R package
“curatedTCGAData” (Ramos et al., 2020). Only pri-
mary solid tumors are considered and technical repli-
cates were no present in each data view. The follow-
ing data sources are considered:
• miRNA gene-level expression values (log2 RPM)
from RNA-sequencing
• mRNA gene expression values (TPM) from RNA-
sequencing
• normalized protein expression values from Re-
verse Phase Protein Array
Moreover, cancer stages were downloaded and the
samples dichotomized into early-stage cancer (stage
I and stage II) and late-stage cancer (stage III and
stage IV) (Dianatinasab et al., 2018). We consid-
ered a common set of 628 patients across data sources
and removed features having missing values in pro-
tein data. In this way, a complete multi-omic dataset
is obtained, where each data view has the same set
of samples. The dataset was further filtered to re-
tain only the most interesting features for each data
source. A feature is removed if it has zero variance or
if it has very few unique values with respect to sam-
ples cardinality and the ratio of the frequency of the
most common value to the frequency of the second
most common value is large (Kuhn, 2021). After min-
max normalization, features were again filtered based
on Pearson correlation. In particular, features with
an absolute correlation higher than 0.75 are selected
and the one with the largest mean absolute correlation
with respect to the other features is removed (Kuhn,
2021). At the end of this filtering stage, we have 494
miRNA, 13220 mRNA and 202 proteins. Moreover,
the complete dataset is randomly amputed removing
10%, 20% and 30% of the features from each data
source. The only constrain is avoiding to remove the
same patient in the third data source if it was already
removed in the other two views. In this way we avoid
to have no data across sources for a specific sample.
At the end, we obtained four different datasets:
1. Complete dataset: no partial samples are present
2. Amputed 10%: each view has 10% of samples
completely missing
3. Amputed 20%: each view has 20% of samples
completely missing
4. Amputed 30%: each view has 30% of samples
completely missing
3.2 Experimental Setup
We aim at comparing the performance of SNF (Wang
et al., 2014) with respect to our proposed approach
miss-SNF. Since SNF can integrate only data sources
without partial samples, we used SNF to integrate the
breast cancer complete dataset and we used miss-SNF
to integrate the amputed datasets. In particular, we
run both the algorithms setting 20 iterations for the
diffusion process and k = 20 for the k- Nearest Neigh-
bours used to build the “local” similarity matrix S (see
equation 4), as suggested by SNF’s authors (Wang
et al., 2021). In both cases, scaled exponential eu-
clidean distance is used to compute the unimodal sim-
ilarity matrices (k = 20, µ = 0.5). The integrated ma-
trices obtained with SNF and miss-SNF are used to
Patient Similarity Networks Integration for Partial Multimodal Datasets
231
perform the prediction of late vs early-stage samples
using a 10 multiple hold-out procedure.
The label propagation algorithm (Zhu et al., 2003)
is exploited to perform a ranking of the predicted late
vs early stage breast cancer patients. A threshold of
0.5 was used to dichotomize the computed normal-
ized scores (scores are normalized between 0 and 1
by min-max normalization) and the following metrics
are computed to evaluate the generalization perfor-
mance on the test set: precision, recall, specificity,
F-measure, accuracy, AUC, AUPRC. Moreover, the
RMSE (Root Mean Squared Error) between the in-
tegrated matrix obtained by SNF (P
(c)
comp
) and the in-
tegrated matrices obtained by miss-SNF on the am-
puted datasets (P
(c)
amp
) is computed, in order to evaluate
whether the proposed algorithm is able to recover the
integrated data set when partial samples are present:
RMSE =
v
u
u
u
t
n
∑
i=1
P
(c)
comp
− P
(c)
amp
2
n
3.3 Experimental Results
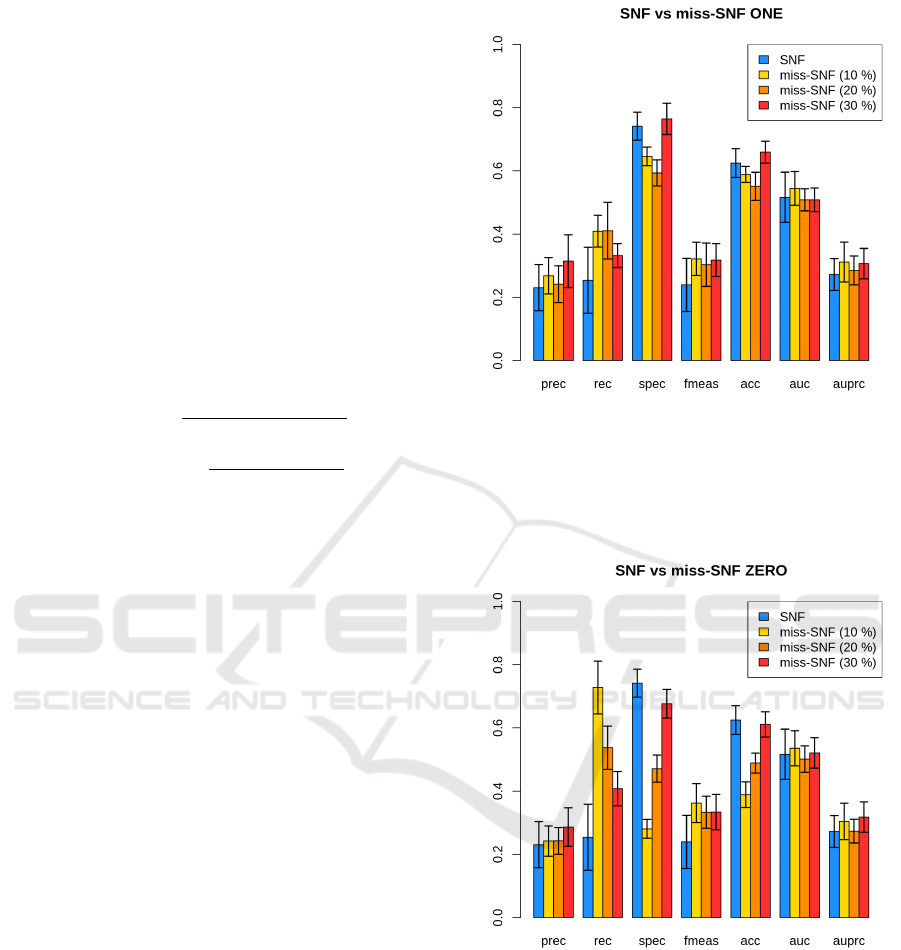
Figure 1 shows the performance of SNF and miss-
SNF ONE on the different amputed datasets, while
Figure 2 shows the performance of miss-SNF ZERO.
In Figure 1, we can see that miss-SNF ONE has com-
petitive results with respect to SNF in terms of pre-
cision, AUC and AUPRC for all the percentages of
dataset amputation, while it is even able to surpris-
ingly improve recall and F-measure. On the other
hand, we can spot a drop in specificity and accuracy
for 10% and 20% of amputation, while the results
seems comparable with miss-SNF when the dataset
has a 30% of missing samples. A similar behaviour
is shown by miss-SNF ZERO even if performance
are generally slightly worse (except for recall and F-
measure) with respect to miss-SNF ONE, probably
due to the missing data reconstruction performed by
the latter. Indeed, Table 1 shows a slightly lower
RMSE for miss-SNF ONE with respect to miss-SNF
ZERO and the difference increases when the percent-
age of missing data is higher, showing that, as ex-
pected, miss-SNF ONE is able to partially recover the
missing samples.
4 DISCUSSION AND
CONCLUSIONS
In this work we presented miss-SNF, a novel algo-
rithm able to integrate PSN built from different bio-
Figure 1: Performance of SNF and miss-SNF ONE on the
different amputed datasets (10%, 20%, 30%). Results are
averaged across multiple holdouts and error bars show stan-
dard error. “Prec” is precision, “Rec” is recall, “Spec” is
specificity, “fmeas” is F-measure, “acc” is accuracy, “auc”
is the Area under the ROC Curve and “auprc” is the Area
under the Precision-Recall Curve.
Figure 2: Performance of SNF and miss-SNF ZERO on the
different amputed datasets (10%, 20%, 30%). Results are
averaged across multiple holdouts and error bars show stan-
dard error. “Prec” is precision, “Rec” is recall, “Spec” is
specificity, “fmeas” is F-measure, “acc” is accuracy, “auc”
is the Area under the ROC Curve and “auprc” is the Area
under the Precision-Recall Curve.
logical data sources having partial samples. The pro-
posed method leverages the non-linear integration ap-
proach based on message-passing theory presented in
SNF (Wang et al., 2014) to fuse multi-modal data and
to partially reconstruct missing data coming from the
presence of partial samples, i.e. samples having one
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
232

Table 1: RMSE of miss-SNF ONE and ZERO for the differ-
ent amputed (Amp.) datasets with respect to SNF applied
on the complete dataset.
Amp. 10% Amp. 20% Amp. 30%
miss-SNF
ONE 0.0020 0.0021 0.0022
miss-SNF
ZERO 0.0022 0.0025 0.0028
or more completely missing data sources. Many ap-
proaches able to integrate PSN computed from differ-
ent sources stem from the algorithm SNF (see (Ma
and Zhang, 2017; Liu and Shang, 2018; Jiang et al.,
2019; Ruan et al., 2019; Rappoport and Shamir, 2019;
Liu et al., 2021; Li et al., 2022; Wu et al., 2021)), but
only NEMO (Rappoport and Shamir, 2019) modified
the original method to take into account the presence
of partial samples, which is a largely overlooked prob-
lem in literature. Of note, NEMO requires that each
pair of patients should have at least one common data
source to be integrated. This assumption is absent in
miss-SNF. To the best of our knowledge, miss-SNF is
the first “SNF-based approch” (Gliozzo et al., 2022)
able to handle partial samples without such constraint.
We showed on a breast cancer multi-omic dataset that
miss-SNF can achieve comparable or even better per-
formance with respect to SNF considering different
percentages of partial samples present in the dataset.
Moreover, we showed that SNF is able to reconstruct
missing data. In future works, we plan to extensively
test miss-SNF on other multi-omics cancer datasets
of different sample size and on non-cancer datasets.
Moreover, we will compare miss-SNF with state-of-
the-art methods able to integrate multiple data sources
and handle the presence of completely missing sam-
ples in the dataset (Rappoport and Shamir, 2019; Xu
et al., 2021).
REFERENCES
Akhoon, N. (2021). Precision medicine: a new paradigm
in therapeutics. International Journal of Preventive
Medicine, 12.
Conesa, A. and Beck, S. (2019). Making multi-omics data
accessible to researchers. Scientific data, 6(1):1–4.
Dianatinasab, M., Mohammadianpanah, M., Daneshi,
N., Zare-Bandamiri, M., Rezaeianzadeh, A., and
Fararouei, M. (2018). Socioeconomic factors, health
behavior, and late-stage diagnosis of breast cancer:
considering the impact of delay in diagnosis. Clini-
cal breast cancer, 18(3):239–245.
Gliozzo, J., Mesiti, M., Notaro, M., Petrini, A., Patak, A.,
Puertas-Gallardo, A., Paccanaro, A., Valentini, G.,
and Casiraghi, E. (2022). Heterogeneous data integra-
tion methods for patient similarity networks. Briefings
in Bioinformatics, 23(4).
Hutter, C. and Zenklusen, J. (2018). The cancer genome
atlas: Creating lasting value beyond its data. Cell,
173(2):283–285.
Jiang, L., Xiao, Y., Ding, Y., Tang, J., and Guo, F. (2019).
Discovering cancer subtypes via an accurate fusion
strategy on multiple profile data. Frontiers in genetics,
10:20.
Kuhn, M. (2021). caret: Classification and Regression
Training. R package version 6.0-90.
Li, L., Wei, Y., Shi, G., Yang, H., Li, Z., Fang, R., Cao,
H., and Cui, Y. (2022). Multi-omics data integra-
tion for subtype identification of chinese lower-grade
gliomas: A joint similarity network fusion approach.
Computational and Structural Biotechnology Journal,
20:3482–3492.
Lightbody, G., Haberland, V., Browne, F., Taggart, L.,
Zheng, H., Parkes, E., and Blayney, J. K. (2019). Re-
view of applications of high-throughput sequencing
in personalized medicine: barriers and facilitators of
future progress in research and clinical application.
Briefings in bioinformatics, 20(5):1795–1811.
Liu, J., Liu, W., Cheng, Y., Ge, S., and Wang, X. (2021).
Similarity network fusion based on random walk
and relative entropy for cancer subtype prediction of
multigenomic data. Scientific Programming, 2021.
Liu, S. and Shang, X. (2018). Hierarchical similarity net-
work fusion for discovering cancer subtypes. In Inter-
national Symposium on Bioinformatics Research and
Applications, pages 125–136. Springer.
Ma, T. and Zhang, A. (2017). Integrate multi-omic data
using affinity network fusion (anf) for cancer patient
clustering. In 2017 IEEE International Conference on
Bioinformatics and Biomedicine (BIBM), pages 398–
403. IEEE.
Pai, S. and Bader, G. D. (2018). Patient similarity networks
for precision medicine. Journal of molecular biology,
430(18):2924–2938.
Ramos, M., Geistlinger, L., Oh, S., Schiffer, L., Azhar, R.,
Kodali, H., de Bruijn, I., Gao, J., Carey, V. J., Mor-
gan, M., and Waldron, L. (2020). Multiomic inte-
gration of public oncology databases in bioconduc-
tor. JCO Clinical Cancer Informatics, (4):958–971.
PMID: 33119407.
Rappoport, N. and Shamir, R. (2019). Nemo: cancer
subtyping by integration of partial multi-omic data.
Bioinformatics, 35(18):3348–3356.
Ruan, P., Wang, Y., Shen, R., and Wang, S. (2019). Us-
ing association signal annotations to boost similarity
network fusion. Bioinformatics, 35(19):3718–3726.
Tomczak, K., Czerwi
´
nska, P., and Wiznerowicz, M. (2015).
Review the cancer genome atlas (tcga): an immea-
surable source of knowledge. Contemporary Oncol-
ogy/Wsp
´
ołczesna Onkologia, 2015(1):68–77.
Wang, B., Mezlini, A., Demir, F., Fiume, M., Tu, Z.,
Brudno, M., Haibe-Kains, B., and Goldenberg, A.
(2021). SNFtool: Similarity Network Fusion. R pack-
age version 2.3.1.
Wang, B., Mezlini, A. M., Demir, F., Fiume, M., Tu, Z.,
Brudno, M., Haibe-Kains, B., and Goldenberg, A.
(2014). Similarity network fusion for aggregating data
Patient Similarity Networks Integration for Partial Multimodal Datasets
233

types on a genomic scale. Nature methods, 11(3):333–
337.
Wu, Y., Wang, H., Li, Z., Cheng, J., Fang, R., Cao, H., and
Cui, Y. (2021). Subtypes identification on heart failure
with preserved ejection fraction via network enhance-
ment fusion using multi-omics data. Computational
and Structural Biotechnology Journal, 19:1567–1578.
Xu, H., Gao, L., Huang, M., and Duan, R. (2021). A net-
work embedding based method for partial multi-omics
integration in cancer subtyping. Methods, 192:67–76.
Zhu, X., Ghahramani, Z., and Lafferty, J. D. (2003). Semi-
supervised learning using gaussian fields and har-
monic functions. In Proceedings of the 20th Inter-
national conference on Machine learning (ICML-03),
pages 912–919.
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
234
