
DT-ML: Drug-Target Metric Learning
Domonkos Pog
´
any
a
and P
´
eter Antal
b
Department of Measurement and Information Systems, Budapest University of Technology and Economics,
Budapest, Hungary
Keywords:
Drug-Target Interaction Prediction, Drug Repositioning, Representation Learning, Metric Learning, Joint
Embedding Models, Negative Sampling.
Abstract:
The challenges of modern drug discovery motivate the use of machine learning-based methods, such as pre-
dicting drug-target interactions or novel indications for already approved drugs to speed up the early discovery
or repositioning process. Publication bias has resulted in a shortage of known negative data points in large-
scale repositioning datasets. However, training a good predictor requires both positive and negative samples.
The problem of negative sampling has also recently been addressed in subfields of machine learning with
utmost importance, namely in representation and metric learning. Although these novel negative sampling
approaches proved to be efficient solutions for learning from imbalanced data sets, they have not yet been
used in repositioning in such a way that the learned similarities give the predicted interactions.
In this paper, we adapt representation learning-inspired methods in pairwise drug-target/drug-disease predic-
tors and propose a modification to one of the loss functions to better manage the uncertainty of negative
samples. We evaluate the methods using benchmark drug discovery and repositioning data sets. Results indi-
cate that interaction prediction with metric learning is superior to previous approaches in highly imbalanced
scenarios, such as drug repositioning.
1 INTRODUCTION
One of the main motivations for modern drug devel-
opment is the discovery of new candidate compounds
which can be used as medication. Developing a new
drug molecule is a long and expensive process; bring-
ing a new drug to market takes approximately 10–15
years and 1.5–2.0 billion USD (Wouters et al., 2020).
One possible way to accelerate the development pro-
cess is via drug repositioning. Repositioning or re-
purposing refers to using a known drug in a new ther-
apeutic application, which is a promising approach,
considered less time-intensive, costly, and risky com-
pared to de novo molecule design.
The different stages of drug development have
been heavily influenced by the rise of artificial in-
telligence technologies in recent years. As a result
of this, classical machine learning methods have be-
come increasingly common among drug-target inter-
action (DTI) prediction approaches (Bagherian et al.,
2021). We can reduce the cost and time required for
measuring the interactions with their help. Besides,
a
https://orcid.org/0000-0003-4968-7504
b
https://orcid.org/0000-0002-4370-2198
these models can later be used to estimate the inter-
action between an unknown protein and molecule, to
search for candidates at the beginning of the devel-
opment process that binds to a specific protein, or to
reveal a new therapeutic application to a known drug,
i.e., repositioning (Harrer et al., 2019).
The simplest methods give estimates based only on
the similarity of molecules (Lee et al., 2016), or treat
the problem as a classification and apply neural net-
works (Arany et al., 2022). Utilizing matrix factoriza-
tion (MF) is a common approach too (Bolg
´
ar and An-
tal, 2017). Still, most of the state-of-the-art (SOTA)
solutions use a general version of MF, namely pair-
wise
1
neural networks, such as the DeepDTA (
¨
Ozt
¨
urk
et al., 2018) or the AI-Bind (Chatterjee et al., 2021).
While the AI-Bind method utilizes pre-trained repre-
sentations, the DeepDTA model uses convolutional
encoders to transform the SMILES representations
from the molecular side and the amino acid sequences
from the protein side, thus providing the latent em-
beddings. These are concatenated, and a multilayer
perceptron (MLP) predicts the interactions. Most
1
Pairwise predictors have dual inputs, for instance, a
molecule and a protein, hence their name.
204
Pogány, D. and Antal, P.
DT-ML: Drug-Target Metric Learning.
DOI: 10.5220/0011691100003414
In Proceedings of the 16th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2023) - Volume 3: BIOINFORMATICS, pages 204-211
ISBN: 978-989-758-631-6; ISSN: 2184-4305
Copyright
c
2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)

of the aforementioned approaches were first applied
in recommendation systems but are now considered
SOTA in the field of DTI prediction too.
In repositioning, the aim is not to estimate a specific
interaction accurately but to establish a good disease
or molecule ordering. Accordingly, several methods
diverge from the traditional approach of treating in-
teraction prediction as a binary classification and ap-
plying new loss functions better suited for ranking,
such as the Bayesian Personalized Ranking (BPR)
loss (Peska et al., 2017), also adopted from the field
of recommendation systems.
Sufficient quality and quantity of data are necessary
to apply a statistical learning approach. There are
plenty of available data sets for DTI prediction tasks,
but due to the high cost of interaction measurements,
the sparsity of these sets is relatively high. Moreover,
the number of known negative entries in drug-disease
interactions is lower than expected due to publica-
tion bias, where negative results are often not pub-
lished (Luo et al., 2021). Therefore, drug-disease
matrices are not only sparse, but often only the posi-
tive entries are known. This is a common problem in
repositioning tasks since SOTA predictors work with
a loss function such as binary cross-entropy (BCE),
which needs the negative samples too. One possible
solution is negative sampling, but since the unknown
entries can be either positive or negative, constructing
a proper sampling method is challenging.
The problem of unknown negative samples has
arisen in representation and (distance) metric learn-
ing too, especially in the field of contrastive learn-
ing (Le-Khac et al., 2020). The main motivation is
to handle a large amount of available unlabeled data
with machine learning. One way to do this is to learn
representations in a self-supervised way. These em-
beddings can later be used in various supervised tasks
if they correctly capture the underlying data distribu-
tion.
Contrastive representation learning is one of the first
widely used solutions, both in the computer vision,
natural language processing, and audio processing
domains (Le-Khac et al., 2020). Architecturally, these
methods can also be classified as pairwise, or rather,
joint embedding methods, because in most cases, one
input pair or triplet is compared at a time. The input
embeddings are first processed by an encoder, thus
creating the latent/metric representations, which are
compared with a similarity function, and finally, a
loss function is used to optimize the similarity be-
tween the pairs. The similarities of positive and neg-
ative pairs are maximized and minimized during op-
timization. Positive pairs can easily be produced with
augmentation, but negative sampling is a challeng-
ing research problem. Unfortunately, using only pos-
itive pairs may lead to a collapse of the representation
space since providing the same embedding for all en-
tries can reduce the loss to zero. Therefore, negative
sampling is necessary for contrastive methods.
Over the last few years, the development of different
contrastive and, later, non-contrastive approaches has
been an area of particular research interest.
The first approaches were the energy-based con-
trastive loss functions, such as the Pair loss (Hadsell
et al., 2006) and the Triplet loss (Collobert and We-
ston, 2008). They aim to associate low energy, i.e.,
low distance, to positive pairs and high energy to neg-
ative pairs.
A new, more effective family of methods is the proba-
bilistic loss functions. Here, a likelihood is described
by a SoftMax function with the similarity to the posi-
tive pair in the denominator and the similarity to all
positive and all negative samples in the numerator.
As opposed to the energy-based methods, it is not the
quality but the quantity of the selected negative sam-
ples that matters since we want to approximate the
denominator as accurately as possible. Therefore, we
often take the samples not only from a single batch
but keep their elements in a so-called memory bank
over several batches. One way of sampling is using
noise contrastive estimation (NCE), and a commonly
used probabilistic loss function is the infoNCE or also
known as normalized-temperature cross-entropy (NT-
Xent) (Chen et al., 2020). A modified version of
NT-Xent is called Supervised contrastive loss (Sup-
Con) (Khosla et al., 2020). The authors of this paper
performed supervised representation learning, where
the labels resulted from a classification problem, and
proposed SupCon, which can handle entities belong-
ing to the same class. Another function is the cir-
cle loss (Sun et al., 2020). The novelty is that it
does not increase the similarity of positive pairs and
decrease the similarity of negative pairs equally but
adaptively assigns different gradient weights. It does
this by defining an optimum for the positive and neg-
ative similarities and then weighting each pair by the
deviation from it.
Because of the efficiency problems associated with
negative sampling, research in recent years has fo-
cused on non-contrastive approaches, which also aim
to avoid latent collapse but do so without nega-
tive samples, for instance, the Variance-Invariance-
Covariance Regularization, (VICReg) (Bardes et al.,
2021).
There are striking similarities between pairwise
DTI prediction methods and joint embedding repre-
sentation learning approaches. Namely, both utilize
two inputs, from which two latent embeddings are
DT-ML: Drug-Target Metric Learning
205

learned, and the output is given by comparing them.
For example, the concatenation and MLP, mentioned
by the DeepDTA and AI-Bind models, can be consid-
ered a special similarity function with trainable pa-
rameters.
This analogy is also true for the MF approaches,
where the similarity function is a simple dot prod-
uct. In the Metric Factorization (Zhang et al., 2018)
collaborative filtering method, the idea appeared that
during matrix factorization, we map users and prod-
ucts into a common space, and the similarity in this
space is the prediction. But this was exploited only
to the extent that instead of scalar multiplication, Eu-
clidean distance was used. The first successful ap-
proach that combines the two is called Collaborative
Metric Learning (CML) (Hsieh et al., 2017), which
uses the Weighted Approximate-Rank Pairwise Loss
(WARP) (Weston et al., 2010). The loss uses triplets
of a user and a positive and negative item and gives
weights to the triplets proportional to the approxi-
mated rank of the positive sampled item in the row
of the given user, i.e., those who are further back in
the row are penalized more. This way, the WARP is
better suited for ranking than the BCE loss, the au-
thors have also tried the BPR ranking-based loss, but
the WARP was found to be superior.
To the best of our knowledge, novel negative sam-
pling solutions developed in the field of metric learn-
ing have not yet been applied to drug repositioning
or DTI prediction this way, namely by treating the
learned similarity as a predicted interaction. How-
ever, we have seen that ideas that have worked in the
collaborative learning field are adopted sooner or later
by interaction prediction methods. This would be par-
ticularly useful for drug repositioning because metric
learning-based approaches provide better solutions to
the problem of negative samples than the current re-
purposing methods, mainly using BCE loss with neg-
ative sampling. Some approaches use BPR, which is
better suited for ranking, but the novel contrastive and
non-contrastive loss functions have not yet been uti-
lized to predict interactions. In current approaches,
representation learning is only used in the pre-training
phase, e.g., to learn node representations on a multi-
modal, heterogeneous knowledge graph. Later these
embeddings are concatenated and used in interaction
prediction tasks with a BCE loss function (Li et al.,
2022), or the adjacency matrix is reconstructed from
them (Chen et al., 2022).
To this end, we propose a drug-Target Metric
Learning (DT-ML) approach that combines the two
methods. In this paper, different metric learning-
based methods are utilized and examined by their ap-
plicability to interaction prediction and drug reposi-
tioning. According to the results, among the various
DT-ML approaches compared, the ones using proba-
bilistic loss functions have proven superior, even bet-
ter than the current SOTA. Additionally, we propose
modifying one of the used loss functions, which could
further improve the results.
2 METHODOLOGY
An overview of the DT-ML methodology is shown in
Figure 1, detailing the data sets, architecture, similar-
ity and loss functions, and metrics used in the evalua-
tion.
2.1 Data and Representations
We utilized two widely used benchmark data sets to
evaluate our models, namely KiBA (Tang et al., 2014)
and ChEMBL (Gaulton et al., 2017). The former is
a DTI data set, with interactions between molecules
and proteins and known negative entries; the latter is
used for repositioning, as it contains drugs with only
positive indications of human conditions, i.e., associ-
ated diseases. In addition, a third data set was used
to produce disease representations, namely the Dis-
GeNET (Pi
˜
nero et al., 2016), which contains relation-
ships between diseases and genes.
The KiBA set contains 467 kinase proteins, and
their interactions with molecules are given with a
dissociation constant (pK
d
). After preprocessing
the compounds, we retained only those for which
the canonical SMILES descriptor is known, unique,
and contains no more than 100 non-hydrogen atoms,
yielding 50,418 molecules. We discretized the inter-
action data with a threshold of pK
d
= 3, as suggested
by the authors of DeepDTA. This resulted in 72,944
positive and 162,681 negative entries; thus, the den-
sity of the interaction matrix is ∼1%.
Among the many tried protein representations, the
512-dimensional CPCProt (Lu et al., 2020) proved to
be the best. On the compound side, the pretrained,
300-dimensional Mol2vec embedding (Jaeger et al.,
2018) gave the best results and was also the most effi-
cient to work with, thus was chosen for all subsequent
work
2
.
Another data set we used is ChEMBL. It contains
drug-like bioactive substances that are already FDA-
approved or are in clinical trials, and associated in-
dications as Medical Subject Headings (MeSH) (Lip-
scomb, 2000). MeSH is a controlled vocabulary of
2
Utilizing Mol2vec on the input is widespread in the
literature, e.g., the AI-Bind model also uses it.
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
206
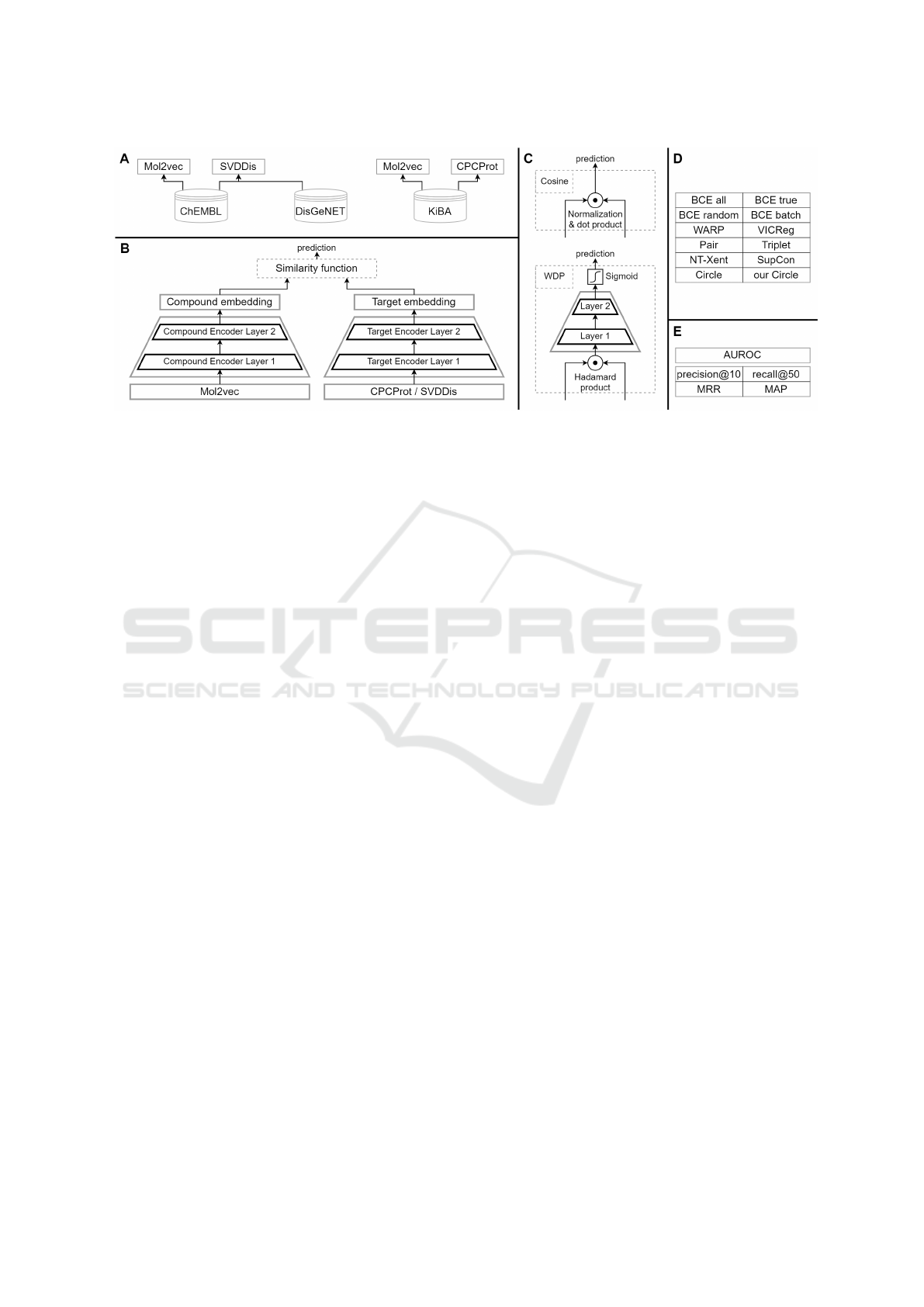
Figure 1: Visual summarization of the DT-ML methodology. (A) Used data sets with different types of molecule, protein, and
disease embeddings. (B) The implemented pairwise architecture. (C) Two used similarity functions in detail. (D) The list of
used loss functions and evaluation metrics (E).
life science concepts and terms also used in the litera-
ture. The part of the data set we use has 21,042 known
positive relationships between 4,755 drugs and 1,168
diseases, resulting in a density of 0.3789%.
To represent molecules, we used the Mol2vec. To ob-
tain disease embeddings, we utilized the DisGeNET
data set, which contains 1,134,924 positive entries
between 30,170 diseases and 21,666 genes, giving
a density of 0.1736%. Although there are no well-
established methods for representing diseases as there
are for proteins, a simple disease embedding can be
easily obtained in a semi-supervised way based on
the known disease-gene associations. We used trun-
cated singular value decomposition (SVD) to con-
vert the 21.666-dimensional sparse vectors into 64-
dimensional dense, gene-based disease representa-
tions, later referred to as SVDDis. MeSH concepts
map diseases from each column in the ChEMBL ma-
trix to a row in DisGeNET, thus we can use SVDDis
embeddings to represent diseases in the repositioning
data set. Another possible option is to use one-hot
representations, but this gave worse results, and with-
out the embeddings, the model is no longer able to
give predictions for new diseases.
2.2 Pairwise DTI Predictor
After preprocessing the data, we implemented a pair-
wise model using the PyTorch package.
We used the previously mentioned Mol2vec,
CPCProt, and SVDDis embeddings as inputs. Af-
ter scaling them on the training data, these embed-
dings are further transformed by two encoders, thus
creating latent representations. We concluded that
the method is less sensitive to the hyperparameters
of the encoders. After trying several combinations,
we finally chose two-layer MLP modules, with 512-
dimensional hidden, and 256-dimensional output lay-
ers. Between layers, a Rectified Linear Unit (ReLU)
activation and 20% dropout rate were used.
We defined fixed and trainable similarity functions
to obtain a prediction of a given interaction from the
metric embeddings and to measure the similarity be-
tween the entities. As a fixed similarity, we have tried
Manhattan, Euclidean, mean squared, dot product,
and cosine similarities, among them, the latter proved
to be superior. Several trainable similarity functions
were tested too. We found that instead of concatena-
tion, it is better to take the Hadamard product and use
an MLP module with a sigmoid activation to obtain
the predictions. We used a multilayer perceptron with
two, 256, and 128-dimensional hidden layers and a
dropout with a rate of 10%. We refer to this later as
the weighted dot product (WDP) similarity.
Figure 1 shows the architecture of the pairwise
model and the used similarity functions. The model
can be used with BCE loss function as a simple pair-
wise DTI predictor, or even with different loss func-
tions according to the DT-ML approach.
2.3 Loss Functions
In our study, we tested several loss functions and neg-
ative sampling strategies.
As a ranking-based baseline, we implemented the
WARP and used it with a margin of 0.1.
In the other baseline approaches, we have used BCE
loss with sampling. The simplest approach is random
sampling, in this case, we tried different ratios, but we
found it best to sample twice as many negative sam-
ples as the number of known positives. This approach
is later referred to as BCE random.
DT-ML: Drug-Target Metric Learning
207

We tested the closed-world assumption, where all the
unknown entries are assumed to be negative, in this
case, the models worked with a fully completed ma-
trix. We refer to this as BCE all.
This method is inefficient to use on the KiBA data
set due to the number of possible interactions. On the
other hand, in the KiBA data set, there are known neg-
ative interactions too, which can be utilized instead of
sampling. In most cases, we discarded the negative
entries of the KiBA set and used negative sampling
just as with the ChEMBL data set so as not to com-
promise comparability, but we kept one case where
we used the known negatives (BCE true).
We have also examined sampling during training,
here, negative samples were given by unknown sam-
ples within a batch (BCE batch).
To improve the results, we weighted the positive and
negative terms in the BCE loss. The weights are in-
versely proportional to the proportion of positive and
negative samples in the data set. So even though there
are more negative samples, they are taken into ac-
count with less weight, this way, we can express the
uncertainty in the noisy negative sampling.
The above-listed baseline approaches represent
the current SOTA, which we compared with several
metric learning-inspired loss functions. Compared to
the previous BCE and WARP functions, one of the
main differences is that DT-ML methods are not only
able to compare molecules with targets, but they also
utilize molecule-molecule and target-target similari-
ties in a semi-supervised way. This way, molecules,
and targets are represented in a common latent space,
and here the same similarity function is applied to
compare the different types of modalities with them-
selves and with each other
3
. During the optimization
of the DT-ML models, only the interacting molecule-
target pairs within a batch are considered positive,
negative pairs are sampled from the various possible
molecule-molecule, molecule-target, and target-target
combinations.
We have tried all loss functions implemented in
the PyTorch Metric Learning framework (Musgrave
et al., 2020). Of the several fixed and learnable simi-
larity functions we tried, cosine proved to be the best
for these approaches.
First, we examined energy-based loss functions, such
as pair and triplet loss. We used them with a margin
of 0.2, which we found to be optimal. The quality
of the negative samples is a significant factor in using
energy-based functions, hence it is important to select
3
One possible hypothesis is that these embeddings carry
information about binding sites, individuals that share a
common or related binding site will be close in the latent
space.
useful samples. With triplet loss, we only use triplets
in which the positive pair has a greater similarity than
the negative, but the difference between them is less
than the predefined margin.
Among the tested probabilistic loss functions, NT-
Xent, SupCon, and Circle losses were in the top three.
For NT-Xent, a temperature hyperparameter of 0.01
was found to be optimal, and a memory bank capa-
ble of containing 512 interactions was used to fur-
ther improve performance. SupCon can handle en-
tities belonging to the same class better than previous
loss functions. Indeed, when considering molecules
as classes, we obtained better results. This means
that targets binding to the same compound are form-
ing positive pairs in the given batch
4
. The tempera-
ture parameter was set to 0.01 for SupCon too. The
best results were obtained with the Circle loss func-
tion. Besides the γ temperature hyperparameter, it has
two optima and two margins for the positive and neg-
ative pairs, but for simplicity, the authors have used
only one m hyperparameter to define them. Over the
various investigated combinations, m = 0.4, γ = 40
proved to be optimal. Because of the uncertainty of
the negative samples, we propose a modified version
where the positive and negative samples have sepa-
rate hyperparameters, m
p
and m
n
, respectively. With
our Circle loss function, we gave negative samples
a softer margin parameter of m
n
= 0.6, and positive
samples a harder margin of m
p
= 0.3, this way, we
were able to achieve further improvements.
Finally, we examined methods that do not require
negative sampling at all, such as VICReg. This
worked best when the weights of the variance, invari-
ance, and covariance loss terms were equal.
2.4 Evaluation Methods
To evaluate the approaches mentioned above, we uti-
lized a row-wise train-test split with 5-fold cross-
validation. This way, the test data matrix contains
only rows/molecules which were not included during
training, but all the columns/targets used in the evalu-
ation were seen in the training data too. We used five
metrics in total to compare the various methods.
One of them is the area under the receiver op-
erating characteristic curve (AUROC), which is fre-
quently used to evaluate binary classification tasks,
4
The intuition behind this is the previously mentioned
binding site analogy, as a protein or the proteins associated
with one disease contain – on average – far more binding
sites than the number of molecule substructures matching
different sites. Thus, there is a high probability that proteins
that share binding molecules will have a common binding
site, so their representations should be similar indeed.
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
208

hence widespread in interaction prediction too. It
only makes sense to use this metric with the KiBA
data set, because the ChEMBL does not have any
known negative entries. We calculated the AUROC
values on the test columns, which had at least 50 pos-
itive and 50 negative entries in the whole KiBA data
set, after that we took the column-wise average.
We also used four ranking-based metrics because,
in repositioning, the order of the predicted interac-
tions matters more than the actual predicted values of
the interactions. To this end, we calculated the aver-
age precision@10 (later referred to as PREC) of rows
in which there were at least ten entries among the test
set, and the mean recall@50 (REC) value over rows
in which there were at least 5 entries. We also used
the Mean Reciprocal Rank (MRR) and the Mean Av-
erage Precision (MAP).
These values were calculated both at row and col-
umn levels. This was necessary, because, on one
hand, most often, we are not looking for diseases for
a known drug, but rather vice versa, and on the other
hand, this way we can better detect overfitting.
3 RESULTS
We ran our models on a 32GB NVIDIA Tesla
V100 GPU. Among the optimization algorithms tried,
Adaptive Moment Estimation (Adam) proved to be
the best, using an L2 weight decay with a weight
of 10
−5
and a learning rate of 5 ∗ 10
−5
. After a
Xavier weight initialization, we trained the models
over 24 epochs in the case of the KiBA, and over 128
epochs in the case of the ChEMBL data set, we used
a batch size of 256 in both cases. Finally, we evalu-
ated the aforementioned approaches according to the
classification-based and the four ranking-based met-
rics.
On the KiBA data set, according to the AUROC
metric, the SOTA BCE true approach outstandingly
outperformed all the other methods, which is not sur-
prising, since it is the only one using the known pos-
itive and negative entries as well. With the WDP and
Cosine similarities, it managed to reach 0.7851 and
0.7391 AUROC respectively, which are the highest
achieved values among all methods for both similar-
ity functions.
Considering the ranking-based metrics, the results
on the KiBA data set are shown in table 1 while the
results on the ChEMBL set can be seen in table 2.
Although BCE loss trained on the known negatives
is still the most suitable for classification, some of
the DT-ML approaches perform better for reposition-
ing
5
. Among them, the energy-based and the non-
contrastive loss functions achieve poor scores, while
the probabilistic methods perform particularly well,
even better than the SOTA.
The column-based metrics are lower on average
because there are much more rows than columns in
the test data. However, these metrics are more rel-
evant, as they can detect overfitting due to the row-
wise train-test split. SOTA approaches using BCE
or WARP loss reach great results in some of the
row-based metrics but perform poorly according to
column-wise evaluation. In the case of WARP, one
possible reason other than the row-wise split is that
it only uses row-wise ranking, thus attending mainly
to the column representations. This way, interactions
with the same target got similar predictions, which is
not a problem considering row-wise evaluation, but
the model is not able to distinguish interactions be-
tween a given target and different molecules. How-
ever, with DT-ML methods, this inequality between
row-, and column-wise evaluations does not apply.
It can be concluded that DT-ML approaches, es-
pecially the ones with a probabilistic loss function,
perform well at both row and column levels. Mostly
column-wise ranking metrics should be considered
when selecting an appropriate method for drug repo-
sitioning, and according to them, Circle loss, or our
modified version of it, performs best.
4 CONCLUSIONS
We have seen the challenges inherent in drug discov-
ery and how deep learning-based interaction predic-
tion and repositioning, can accelerate the develop-
ment process. Most of the SOTA repositioning ap-
proaches utilize a DTI predictor, which needs both
positive and negative entries to train. However, neg-
ative results are often not published, thus there is a
shortage of negative samples among drug-disease in-
teractions. We have also seen that in recent times
negative sampling has been the main challenge in a
subfield of machine learning with utmost importance,
namely in metric learning too, and the attention in-
vested in researching this area has led to a number of
effective solutions.
5
There is a slight imbalance in the comparability of
models. Much more iterations were performed during one
epoch for the BCE all approach, and methods using the
weighted similarity module have more trainable parame-
ters. In these cases, baseline methods are better according
to some row-based metrics. However, similar performance
can also be achieved by using DT-ML methods with more
parameters or more epochs.
DT-ML: Drug-Target Metric Learning
209
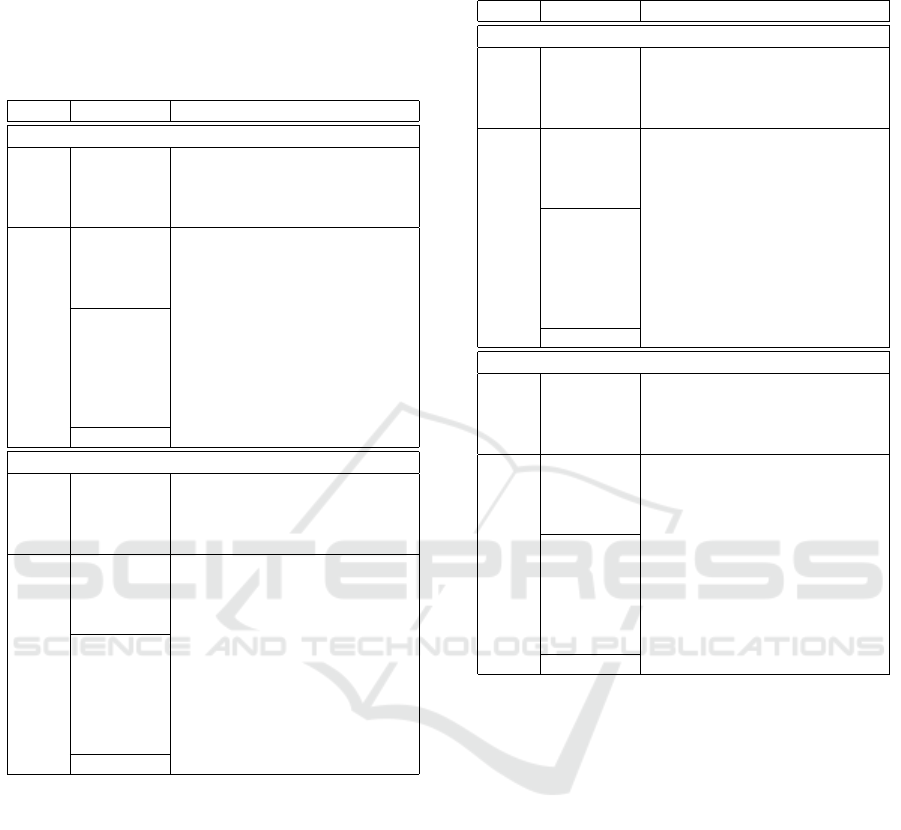
Table 1: Row- and column-wise results on the KiBA data
set, the best two methods are highlighted for each ranking-
based metric. The first four rows contain the baseline,
SOTA methods trained with the WDP similarity module,
below them, there are the baseline and DT-ML approaches
with our modified Circle loss at the bottom, with these
methods, the cosine similarity was used.
Sim. Loss function PREC REC MRR MAP
Column-wise ranking
WDP
BCE true 0.1512 0.0936 0.1437 0.0487
BCE random 0.2866 0.1925 0.2389 0.1218
BCE batch 0.2674 0.1826 0.2497 0.1048
WARP 0.2279 0.1617 0.1931 0.0907
Cosine
BCE true 0.1023 0.0459 0.0893 0.0235
BCE random 0.1273 0.0857 0.1222 0.0448
BCE batch 0.1494 0.0893 0.1642 0.048
WARP 0.3355 0.2039 0.2866 0.1274
Pair 0.0308 0.0264 0.0402 0.0134
Triplet 0.3047 0.1546 0.2897 0.0971
Circle 0.4488 0.2274 0.3614 0.1545
NT-Xent 0.4238 0.2132 0.3392 0.1378
SupCon 0.4244 0.2063 0.3464 0.1393
VICReg 0.043 0.0297 0.0576 0.0164
our Circle 0.461 0.2307 0.3521 0.1536
Row-wise ranking
WDP
BCE true 0.326 0.4026 0.3411 0.3025
BCE random 0.407 0.5664 0.5675 0.53
BCE batch 0.465 0.5454 0.5562 0.5163
WARP 0.447 0.5219 0.554 0.5148
Cosine
BCE true 0.232 0.355 0.1944 0.1684
BCE random 0.316 0.4441 0.3226 0.2823
BCE batch 0.346 0.4774 0.3266 0.2899
WARP 0.433 0.5011 0.5677 0.5223
Pair 0.137 0.2546 0.0448 0.0354
Triplet 0.216 0.3026 0.3377 0.2916
Circle 0.416 0.5584 0.5995 0.5573
NT-Xent 0.37 0.4971 0.5767 0.5325
SupCon 0.408 0.5021 0.5914 0.5491
VICReg 0.344 0.4978 0.4513 0.4112
our Circle 0.429 0.5401 0.6112 0.5669
The major contribution of this study is using these
novel, metric learning-inspired approaches as pair-
wise DTI predictors in the domain of drug reposition-
ing. We showed that DT-ML methods, which to the
best of our knowledge have not yet been applied in
this way, have performed particularly well according
to the ranking metrics, not only at the row but also at
the column level. And finally, we proposed a modi-
fication to the Circle loss to better manage the uncer-
tainty of negative samples.
However, further research is needed, these meth-
ods need to be investigated more in-depth, and other
modifications could be applied. One such possible
improvement is to make better use of the intrinsically
semi-supervised nature of the approach. Molecules
and targets can be augmented within a batch and com-
pared to themselves, thus forming more positive pairs,
Table 2: Row- and column-wise results on the ChEMBL
data set with the best two methods highlighted.
Sim. Loss function PREC REC MRR MAP
Column-wise ranking
WDP
BCE all 0.2064 0.2609 0.2125 0.0998
BCE random 0.1321 0.1874 0.1405 0.0668
BCE batch 0.1893 0.2467 0.1906 0.0893
WARP 0.1421 0.2182 0.1221 0.0621
Cosine
BCE all 0.1229 0.1815 0.1277 0.0533
BCE random 0.1121 0.1671 0.1155 0.0488
BCE batch 0.1121 0.1583 0.1007 0.0439
WARP 0.2171 0.2671 0.2013 0.0874
Pair 0.0357 0.0828 0.0498 0.0214
Triplet 0.2057 0.2304 0.224 0.0945
Circle 0.24 0.2578 0.2198 0.0848
NT-Xent 0.2436 0.2593 0.2424 0.0921
SupCon 0.2486 0.2658 0.2182 0.0842
VICReg 0.0379 0.0924 0.0352 0.0197
our Circle 0.2493 0.2688 0.2315 0.0977
Row-wise ranking
WDP
BCE all 0.3608 0.426 0.3917 0.2374
BCE random 0.3092 0.3839 0.3104 0.1591
BCE batch 0.3105 0.3542 0.3467 0.175
WARP 0.2693 0.3214 0.3099 0.1589
Cosine
BCE all 0.2915 0.3777 0.3322 0.1744
BCE random 0.2719 0.3642 0.3225 0.1778
BCE batch 0.2275 0.3137 0.2046 0.104
WARP 0.2699 0.3229 0.3115 0.1674
Pair 0.0542 0.099 0.0689 0.0251
Triplet 0.2386 0.2967 0.2607 0.131
Circle 0.3725 0.4067 0.4434 0.2591
NT-Xent 0.3373 0.355 0.4187 0.232
SupCon 0.3438 0.3651 0.4483 0.2508
VICReg 0.1837 0.2683 0.2428 0.1418
our Circle 0.3582 0.3926 0.4225 0.2453
and making the representations less sensitive to vari-
ous augmentations. Another promising modification
is to replace the cosine similarity with a trainable
module or even try a hyperbolic embedding space and
similarities developed for non-Euclidean spaces.
ACKNOWLEDGEMENT
This research was funded by the J. Heim Stu-
dent Scholarship (D.P), National Research, Develop-
ment, and Innovation Fund of Hungary under Grant
TKP2021-EGA-02, the OTKA-K139330, the Euro-
pean Union project RRF-2.3.1-21-2022-00004 within
the framework of the Artificial Intelligence National
Laboratory.
REFERENCES
Arany, A., Simm, J., Oldenhof, M., and Moreau, Y.
(2022). Sparsechem: Fast and accurate machine
BIOINFORMATICS 2023 - 14th International Conference on Bioinformatics Models, Methods and Algorithms
210

learning model for small molecules. arXiv preprint
arXiv:2203.04676.
Bagherian, M., Sabeti, E., Wang, K., Sartor, M. A.,
Nikolovska-Coleska, Z., and Najarian, K. (2021). Ma-
chine learning approaches and databases for predic-
tion of drug–target interaction: a survey paper. Brief-
ings in bioinformatics, 22(1):247–269.
Bardes, A., Ponce, J., and LeCun, Y. (2021). Vi-
creg: Variance-invariance-covariance regulariza-
tion for self-supervised learning. arXiv preprint
arXiv:2105.04906.
Bolg
´
ar, B. and Antal, P. (2017). Vb-mk-lmf: fusion
of drugs, targets and interactions using variational
bayesian multiple kernel logistic matrix factorization.
BMC bioinformatics, 18(1):1–18.
Chatterjee, A., Ahmed, O. S., Walters, R., Shafi, Z., Gysi,
D., Yu, R., Eliassi-Rad, T., Barab
´
asi, A.-L., and
Menichetti, G. (2021). Ai-bind: Improving binding
predictions for novel protein targets and ligands. arXiv
preprint arXiv:2112.13168.
Chen, J., Zhang, L., Cheng, K., Jin, B., Lu, X., and
Che, C. (2022). Predicting drug-target interaction via
self-supervised learning. IEEE/ACM Transactions on
Computational Biology and Bioinformatics.
Chen, T., Kornblith, S., Norouzi, M., and Hinton, G. (2020).
A simple framework for contrastive learning of visual
representations. In International conference on ma-
chine learning, pages 1597–1607. PMLR.
Collobert, R. and Weston, J. (2008). A unified architec-
ture for natural language processing: Deep neural net-
works with multitask learning. In Proceedings of the
25th international conference on Machine learning,
pages 160–167.
Gaulton, A., Hersey, A., Nowotka, M., Bento, A. P., Cham-
bers, J., Mendez, D., Mutowo, P., Atkinson, F., Bel-
lis, L. J., Cibri
´
an-Uhalte, E., et al. (2017). The
chembl database in 2017. Nucleic acids research,
45(D1):D945–D954.
Hadsell, R., Chopra, S., and LeCun, Y. (2006). Dimen-
sionality reduction by learning an invariant mapping.
In 2006 IEEE Computer Society Conference on Com-
puter Vision and Pattern Recognition (CVPR’06), vol-
ume 2, pages 1735–1742. IEEE.
Harrer, S., Shah, P., Antony, B., and Hu, J. (2019). Arti-
ficial intelligence for clinical trial design. Trends in
pharmacological sciences, 40(8):577–591.
Hsieh, C.-K., Yang, L., Cui, Y., Lin, T.-Y., Belongie, S.,
and Estrin, D. (2017). Collaborative metric learning.
In Proceedings of the 26th international conference on
world wide web, pages 193–201.
Jaeger, S., Fulle, S., and Turk, S. (2018). Mol2vec: un-
supervised machine learning approach with chemical
intuition. Journal of chemical information and mod-
eling, 58(1):27–35.
Khosla, P., Teterwak, P., Wang, C., Sarna, A., Tian, Y.,
Isola, P., Maschinot, A., Liu, C., and Krishnan, D.
(2020). Supervised contrastive learning. Advances
in Neural Information Processing Systems, 33:18661–
18673.
Le-Khac, P. H., Healy, G., and Smeaton, A. F. (2020). Con-
trastive representation learning: A framework and re-
view. IEEE Access, 8:193907–193934.
Lee, A. A., Brenner, M. P., and Colwell, L. J. (2016). Pre-
dicting protein–ligand affinity with a random matrix
framework. Proceedings of the National Academy of
Sciences, 113(48):13564–13569.
Li, Y., Qiao, G., Gao, X., and Wang, G. (2022). Supervised
graph co-contrastive learning for drug–target interac-
tion prediction. Bioinformatics, 38(10):2847–2854.
Lipscomb, C. E. (2000). Medical subject headings
(mesh). Bulletin of the Medical Library Association,
88(3):265.
Lu, A. X., Zhang, H., Ghassemi, M., and Moses, A.
(2020). Self-supervised contrastive learning of protein
representations by mutual information maximization.
BioRxiv.
Luo, H., Li, M., Yang, M., Wu, F.-X., Li, Y., and Wang,
J. (2021). Biomedical data and computational mod-
els for drug repositioning: a comprehensive review.
Briefings in bioinformatics, 22(2):1604–1619.
Musgrave, K., Belongie, S., and Lim, S.-N. (2020). Pytorch
metric learning. arXiv preprint arXiv:2008.09164.
¨
Ozt
¨
urk, H.,
¨
Ozg
¨
ur, A., and Ozkirimli, E. (2018). Deepdta:
deep drug–target binding affinity prediction. Bioinfor-
matics, 34(17):i821–i829.
Peska, L., Buza, K., and Koller, J. (2017). Drug-target inter-
action prediction: a bayesian ranking approach. Com-
puter methods and programs in biomedicine, 152:15–
21.
Pi
˜
nero, J., Bravo,
`
A., Queralt-Rosinach, N., Guti
´
errez-
Sacrist
´
an, A., Deu-Pons, J., Centeno, E., Garc
´
ıa-
Garc
´
ıa, J., Sanz, F., and Furlong, L. I. (2016). Dis-
genet: a comprehensive platform integrating informa-
tion on human disease-associated genes and variants.
Nucleic acids research, page gkw943.
Sun, Y., Cheng, C., Zhang, Y., Zhang, C., Zheng, L., Wang,
Z., and Wei, Y. (2020). Circle loss: A unified perspec-
tive of pair similarity optimization. In Proceedings
of the IEEE/CVF Conference on Computer Vision and
Pattern Recognition, pages 6398–6407.
Tang, J., Szwajda, A., Shakyawar, S., Xu, T., Hintsanen, P.,
Wennerberg, K., and Aittokallio, T. (2014). Making
sense of large-scale kinase inhibitor bioactivity data
sets: a comparative and integrative analysis. Journal
of Chemical Information and Modeling, 54(3):735–
743.
Weston, J., Bengio, S., and Usunier, N. (2010). Large scale
image annotation: learning to rank with joint word-
image embeddings. Machine learning, 81(1):21–35.
Wouters, O. J., McKee, M., and Luyten, J. (2020). Esti-
mated research and development investment needed
to bring a new medicine to market, 2009-2018. Jama,
323(9):844–853.
Zhang, S., Yao, L., Tay, Y., Xu, X., Zhang, X., and
Zhu, L. (2018). Metric factorization: Recommen-
dation beyond matrix factorization. arXiv preprint
arXiv:1802.04606.
DT-ML: Drug-Target Metric Learning
211
