
A Sequence-Motif Based Approach to Protein Function Prediction via
Deep-CNN Architecture
Vikash Kumar
1
, Ashish Ranjan
2
, Deng Cao
3
, Gopalakrishnan Krishnasamy
3
and Akshay Deepak
1
1
National Institute of Technology Patna, Patna, India
2
ITER, Siksha ’O’ Anusandhan Deemed to be University, Bhubaneswar, India
3
Associate Professor, Department of Mathematics & Computer Science, Central State University, Wilberforce, Ohio, U.S.A.
Keywords:
Protein Sequence, Convolutional Neural Network, Protein Sub-Sequence, Consistency Factor.
Abstract:
The challenge of determining protein functions, inferred from the study of protein sub-sequences, is a complex
problem. Also, a little literature is evident in this regard, while a broad coverage of the literature shows a bias in
the existing approaches for the full-length protein sequences. In this paper, a CNN-based architecture is intro-
duced to detect motif information from the sub-sequence and predict its function. Later, functional inference
for sub-sequences is used to facilitate the functional annotation of the full-length protein sequence. The results
for the proposed approach demonstrate a great future ahead for further exploration of sub-sequence based pro-
tein studies. Comparisons with the ProtVecGen-Plus – a (multi-segment + LSTM) approach – demonstrate,
an improvement of +1.24% and +4.66% for the biological process (BP) and molecular function (MF) sub-
ontologies, respectively. Next, the proposed method outperformed the hybrid ProtVecGen-Plus + MLDA by a
margin of +3.45% for the MF dataset, while raked second for the BP dataset. Overall, the proposed method
produced better results for significantly large protein sequences (having sequence length > 500 amino acids).
1 INTRODUCTION
The study of the role of proteins in (i) the disease
Pathobiology, (ii) the examination of meta-genomes,
and (iii) the discovery of therapeutic targets, are im-
portant tasks that require deep knowledge about the
functions of proteins. In this regard, the functional
knowledge acquisition about proteins is well sup-
ported by the computational approaches that are fast
and economical, though, still needing a good amount
of effort to compete with the evolving dynamics of
proteins – only less than 1% of proteins have reviewed
annotations
1
. The recent trend to infer protein func-
tion(s) show a biasness of the existing works for pro-
tein sequences (Jiang et al., 2016), (Kumari et al.,
2019), (Radivojac et al., 2013), (Fa et al., 2018), (Kul-
manov and Hoehndorf, 2020), (Makrodimitris et al.,
2019), (Ranjan et al., 2019), (Ranjan et al., 2021)
– mostly due to their large and cheap availability,
though works based on protein structures (Yang et al.,
2015), (Gligorijevi
´
c et al., 2021), protein interaction
network (Kulmanov et al., 2018), and others (You
1
This statistics is based on the information from the
UniProtKB (Consortium, 2015).
et al., 2018) are also available.
Protein sequences encode vital patterns, which are
formed due to interactions among amino acids that
in turn fold into proteins’ sub-structures, for exam-
ple, binding sites, to perform the function. This jus-
tifies the necessity for the sub-sequence based ap-
proaches, while the existing approaches are primar-
ily focused on full-length protein sequences (Cao
et al., 2017), (Kulmanov et al., 2018), (Kulmanov and
Hoehndorf, 2020) which makes the function predic-
tion a little less effective. There exist only a few no-
table works (Ranjan et al., 2019), (Ranjan et al., 2021)
that have demonstrated the utility of a sub-sequence-
based methodology. In (Ranjan et al., 2019), the
proposed solution is a (multi-segmentation + LSTM)
based framework. The other work (Ranjan et al.,
2021) is an ensemble (multi-segmentation + tf-idf +
MLDA) method. Both works involve utilizing pre-
dicted function(s) for protein sub-sequences to infer
the function(s) of the full-length protein sequence.
Convolutional neural networks (CNNs) have re-
cently gained popularity as a strong alternative to re-
current neural networks (RNNs), automating feature
representations for biological sequences, and for a va-
riety of tasks such as function prediction (Kulmanov
Kumar, V., Ranjan, A., Cao, D., Krishnasamy, G. and Deepak, A.
A Sequence-Motif Based Approach to Protein Function Prediction via Deep-CNN Architecture.
DOI: 10.5220/0011647800003393
In Proceedings of the 15th International Conference on Agents and Artificial Intelligence (ICAART 2023) - Volume 3, pages 243-251
ISBN: 978-989-758-623-1; ISSN: 2184-433X
Copyright
c
2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)
243
et al., 2018), (Kulmanov and Hoehndorf, 2020), drug-
target prediction (
¨
Ozt
¨
urk et al., 2018), (
¨
Ozt
¨
urk et al.,
2019), etc. In, (Kulmanov et al., 2018) and (Kul-
manov and Hoehndorf, 2020), they applied CNNs for
the complete protein sequences to infer protein func-
tion(s). This paper proposes a framework that uses a
deep CNN-based architecture to first infer the func-
tion(s) of protein sub-sequences and then uses the
inferred function(s) for protein sub-sequences to de-
termine the function(s) of the full-length protein se-
quence. The proposed CNN-based architecture ex-
tracts motif information from the sub-sequence, and
uses it to predict the GO-term(s) for the protein sub-
sequence.
The evaluations of the proposed framework con-
ducted for two independent datasets – biological
process (BP) and molecular function (MF) sub-
ontologies
2
– demonstrated a significant effort of
the proposed framework. The overall improve-
ments with respect to the similar multi-segment based
ProtVecGen-Plus (Ranjan et al., 2019), based on
RNNs, i.e., LSTM network, demonstrated improve-
ments of: +1.24% for the BP dataset and +4.66% for
the MF dataset. Further, when compared to the hy-
brid, ProtVecGen-Plus + MLDA (Ranjan et al., 2019)
method, the proposed work produces improvement of
+3.45% for the MF dataset, while ranked second for
the BP dataset. The proposed method showed bet-
ter results for handling the longer protein sequences
(having sequence length > 500 amino-acids).
Following is the organization of the paper: Sec-
tion 2 is an elaboration of the dataset used for the ex-
periments and the proposed methods. Following this
is a Section 3 for the results discussion. Lastly, the
Section 4 is a conclusion.
2 DATASETS AND METHODS
Here, we will discuss the experimental datasets, the
steps for the segmented dataset construction, and the
proposed method.
2.1 Datasets
Experiments were conducted for two datasets, – cor-
responding to the biological process (BP) and molec-
ular function (MF) sub-ontologies as defined by the
Gene Ontology (GO) (Ashburner et al., 2000). These
datasets were created by downloading reviewed pro-
tein sequences and their mapped functional annota-
2
defined by the Gene Ontology Consortium (Ashburner
et al., 2000).
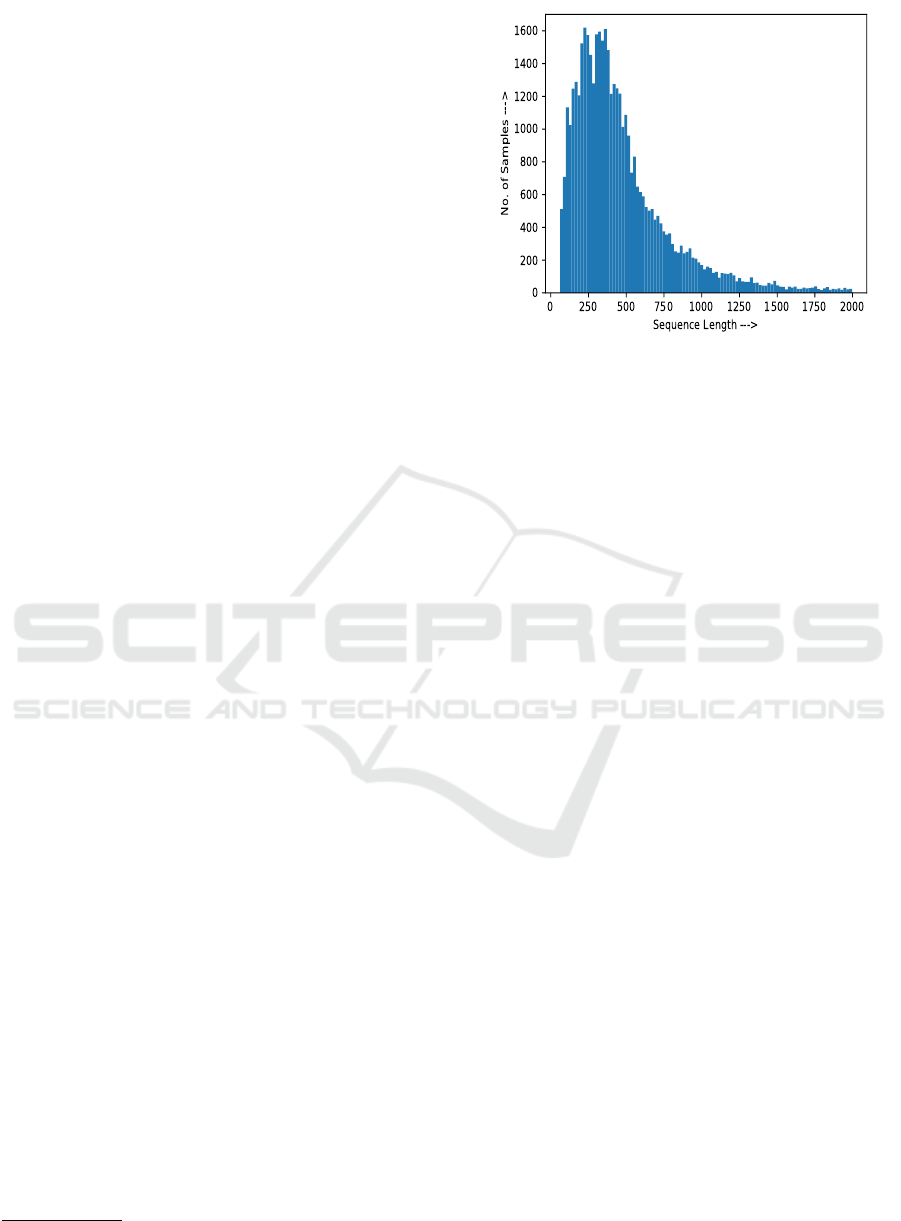
Figure 1: Protein sequences’ distributions are shown.
tions from the UniProtKB/SwissProt (2017) reposi-
tory (Consortium, 2015). The functional annotation
is a unique identifier, known as a GO-term, that in-
dicates the distinct protein function. The biological
process dataset has 58,310 protein sequences and 295
unique GO-terms. The other dataset, molecular func-
tion, has 43,218 protein sequences and 135 unique
GO-terms. For each GO-terms, the least number of
protein sequences is taken as 200. Only proteins with
a sequence length higher than 40 and lower than 2,000
were chosen for this study.
2.2 Steps to Construct the Protein
Sub-Sequence Dataset
The proposed framework to infer the protein func-
tion(s) is based on the protein sub-sequences, hence,
the steps taken to construct the segmented dataset are
explained next. Let the training dataset be denoted
as S = [s
i
,Y
i
]
n
i=1
; where, s
i
and Y
i
denote the i
th
pro-
tein sequence and the corresponding GO-term(s), re-
spectively. As shown in Figure 1, for both the BP
and MF sub-ontologies, large number of protein se-
quences have lengths of around 200 to 300. So, the
maximum length for the protein sub-sequences is set
to 200, with a gap of 60 amino acids between two
consecutive sub-sequences.
1. For each protein sample pair, (s
i
,Y
i
); i ∈
{1, 2,..., n}, a protein sequence s
i
is split to gen-
erate a set of protein sub-sequences of size 200.
Zero-padding is done for the short protein sub-
sequences.
2. The output labels for each sub-sequences are as-
sumed equivalent to the parent protein sequence.
ICAART 2023 - 15th International Conference on Agents and Artificial Intelligence
244
One-Hot Encoding
(dim = 21)
CNN Block 1
Filter Size : 1 X 5
# Filter : 96
CNN Block 2
Filter Size : 1 X 7
# Filter : 96
CNN Block
3,1
Filter Size: 1 X 9
# Filters: 96
CNN Block
3,2
Filter Size: 1 X 9
# Filters: 96
CNN Block
3,N
Filter Size: 1 X 9
# Filters: 96
MaxPooling 1
Flatten 1 Flatten 2 Flatten N
Concatenate
Dropout (0.4)
Output Layer
MaxPooling N
. . . . .
. . . . .
. . . . .
. . . . .
MaxPooling 2
CNN Block
4,1
Filter Size: 1 X 6
# Filters: 128
CNN Block
4,2
Filter Size: 1 X 9
# Filters: 128
CNN Block
4,N
Filter Size: 1 X 27
# Filters: 128
1D CNN
Batch-
Normalization
LeakyRelu
CNN Block
Protein Sequence
Sub-
Network 1
Sub-
Network N
Sub-
Network 2
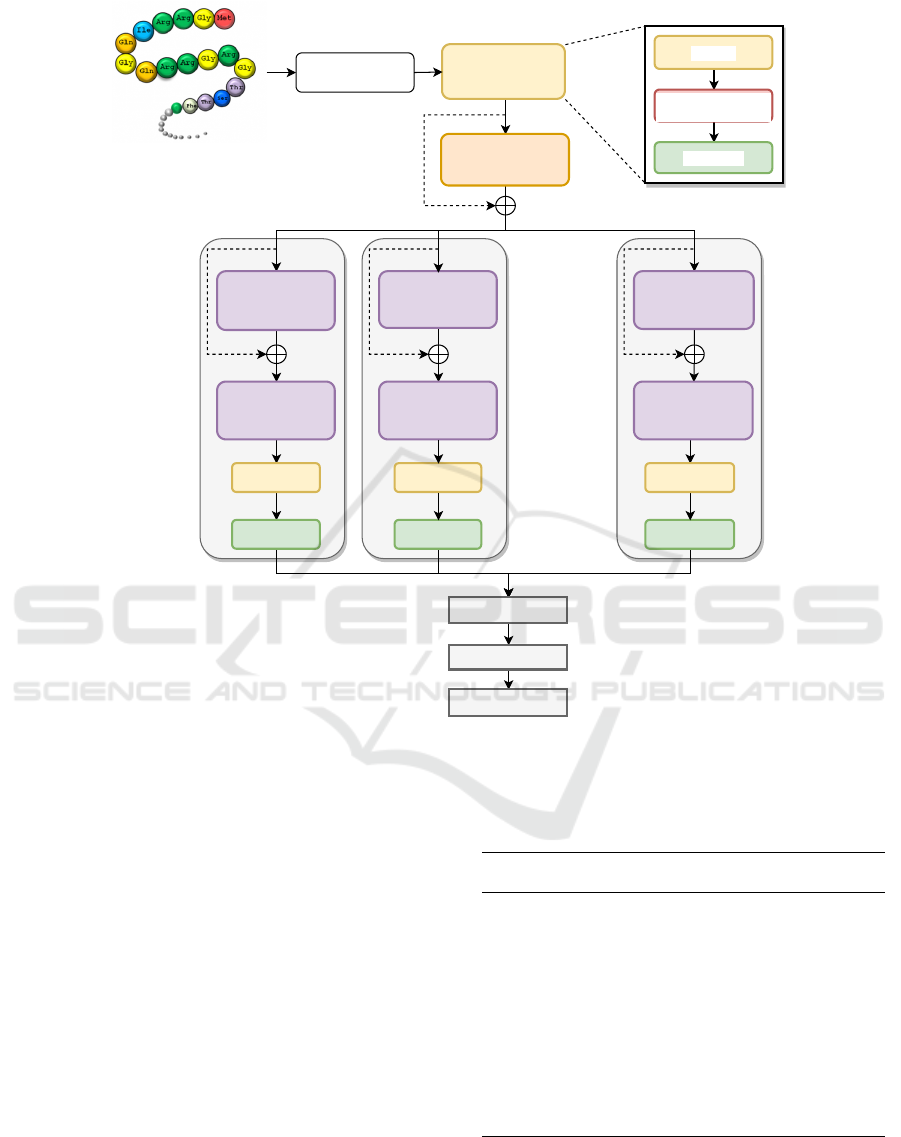
Figure 2: Proposed model architecture with number of sub-networks, denoted as N = 8. The dashed lines indicate the residual
connections.
2.3 Proposed Method
The paper introduces a two-step framework for infer-
ring protein function: (i) a deep stacked CNN-based
architecture is used to first infer function(s) of pro-
tein sub-sequences, and (ii) the inferred function(s)
for protein sub-sequences are used to determine func-
tion(s) of the full-length protein sequence. A discus-
sion on the input sequence representation and the pro-
posed architecture are given next.
2.3.1 One-Hot Encoding Based Input Sequence
Representation
The protein sequences are pre-processed and repre-
sented as a string of amino acids, where amino acids
are represented via the one-hot encoding scheme. The
dimension for the one-hot encoding scheme is taken
as 21. The first dimension is used to indicate the
rare amino acids (O, U, X), whereas the remaining 20
Table 1: Hyper-parameters configurations with different
CNN blocks.
S.
No.
CNN
Block
Filter-
Size
Filters
Count
Note
1. CNN
Block
1
1 x 5 96 –
2. CNN
Block
2
1 x 7 96 –
3. CNN
Block
3
1 x 9 96 –
4. CNN
Block
4
1 x F
s
128 F
s
denote filter size
in range 6, 9, 12, ...,
(3N + 3).
dimensions correspond to the well-known 20 amino
acids.
A Sequence-Motif Based Approach to Protein Function Prediction via Deep-CNN Architecture
245

2.3.2 Stacked CNN-Based Architecture
The proposed architecture employs stacked layers of
Convolutional Neural Networks (CNNs) blocks to
process protein sub-sequences character-by-character
(where amino acids represent a character). The most
notable feature of CNNs is their ability to capture lo-
cal dependencies – between amino acids – through
the use of trainable filters that aid in the transfor-
mation of protein sequence into a viable representa-
tion. The complete architecture is shown in Figure 2.
There are two consecutive layers of two CNN blocks
(CNN Block 1 and CNN Block 2) that are further di-
vided into a set of sub-networks before being com-
bined down the architecture. The components of both
CNN blocks and sub-networks are discussed next:
2.1) CNN Block: The CNN block as shown in
Figure 2 has following layers.
1. 1D-CNN Layer: The purpose of the 1-
dimensional CNN layer is to learn local de-
pendencies between the amino acids along the
sequence. Here, the hyper-parameters that are
customized for different CNN blocks include
the number of filters and filter-size. A more
detailed discussion on these hyper-parameters
with different CNN blocks is given in Table 1. Let
I
f
[n] denote the output feature map after applying
the convolution operation, and the equation can
be seen as:
I
f
[n] = x[n] ∗ h[n] =
∞
∑
−∞
x[k] · h[n − k] (1)
where,
– h[n] is the kernel
– x[n] is the input feature
– * denotes the convolution operation.
2. Batch-Normalization Layer: This layer acts as a
regularizer that controls the biasness of the model
– utilizing the statistics of the mini-batch (Ioffe
and Szegedy, 2015).
Let I
f
denote a input feature-map corresponding
to the f
th
filter, where 1 ≤ f ≤ 128 (given in Table
1), then the output of batch-normalization layer
for the f
th
input feature-map, denoted as I
0
f
, is de-
fined as:
I
0
f
= γ
f
·
I
f
− m[I
f
]
p
var[I
f
]
!
+ β
f
(2)
where,
– I
f
is the f
th
feature map,
– m[I
f
] is the mean of the f
th
feature-map,
– var[I
f
] is the variance for the f
th
feature-map,
– γ
f
and β
f
are two learning parameters which
control m[I
f
] and var[I
f
], respectively.
3. LeakyRelu Layer: This layer transforms the out-
put of the previous layer in the range as given
in Equation 3 and saves the unit from being non-
functional (Maas et al., 2013).
f (I
f
) =
(
αI
f
, if I
f
< 0
I
f
, otherwise
(3)
where,
– α is the constant taken as 0.2.
– I
f
is the given input to the Leaky Relu layer.
2.2) Components of Sub-Network: Each sub-
network is composed of layers as follows:
1. A consecutive layers of two CNN blocks (rep-
resented as CNN Block
3,N
and CNN Block
4,N
).
Here N represents the number of sub-networks.
Different-sized filters with the CNN Block
4,N
are
used to extract motifs of different sizes as given in
Table 1.
2. Next, the MaxPooling layer is employed to ex-
tract features that emphasize the important motifs
present in protein sub-sequences. This also helps
to prevent the over-fitting of the model by reduc-
ing the feature maps.
I
0
f
=
(I
f
− k)
s + 1
(4)
where,
– I
0
f
is the output feature vector.
– I
f
is the input feature vector.
– k is the kernel size
– s is the stride.
3. The last layer of each sub-network is the flatten
layer to reduce the output to a 1-dimensional vec-
tor.
The combined output from each sub-networks,
obtained using the concatenate layer, is then passed to
the dropout layer (with dropout probability = 0.4). Fi-
nally, the output layer with sigmoid activation is used
as a classification layer.
Importantly, a residual connection (shown with
dashed line) is added between the CNN blocks for ef-
ficient training of deep neural architecture, as shown
in Figure 2. This provides a significant improvement
in the network’s ability to overcome vanishing gradi-
ents. The hyper-parameters for the proposed architec-
ture are shown in Table 2.
2.3.3 Final Prediction for the Full-Length
Protein Sequence
The mean of inferred protein functions obtained for
all the protein sub-sequences is computed to get the
ICAART 2023 - 15th International Conference on Agents and Artificial Intelligence
246

Table 2: Hyper-parameters configurations.
S.
No.
Hyper-
Parameters
Values
1. Optimizer Adam (Kingma
and Ba, 2014)
2. Loss Function Binary Cross-
Entropy
3. Learning Rate 5e
−4
4. Clip Value 5.0
final labels for the full-length protein sequence.
3 RESULTS AND DISCUSSION
We have trained this model using Keras built on
the Tensor-flow architecture as the backend. The
datasets discussed in Section 2.1 were split into train-
ing (0.75%) and testing (0.25%) datasets. For mon-
itoring the training, we have used check-pointers,
early-stopping criteria along with 10% of training
data as validation data.
3.1 Evaluation Metrics
Here, let Y
i
= {y
i1
, y
i2
, ...} denotes the actual GO-
terms and P
i
= {p
i1
, p
i2
, ...} denotes the predicted GO-
terms for protein sequence S
i
;i ∈ {1, 2, .., n}. The
metrics were defined as:
1. Average Recall: Recall catches the true prediction
made by the model over all the predicted true sam-
ples.
Re
avg
=
n
∑
i=0
Y
i
∩ P
i
Y
i
(5)
2. Average Precision: Precision catches the true pre-
diction made by the model over all the actual true
samples.
Pr
avg
=
m
∑
i=0
Y
i
∩ P
i
P
i
(6)
3. Average F1-Score: F1-Score balance both the pre-
cision & recall and return the value lowest be-
tween the recall and precision.
F1
avg
=
n
∑
i=0
2|Y
i
∩ P
i
|
|Y
i
| + |P
i
|
(7)
4. Consistency f actor: This is based on variance
which emphasizes a method’s overall general-
ity with regard to protein sequences of various
lengths. This is defined as follows:
Consistency f actor =
r
1
4
∑
( f 1 −
¯
f 1
ri
)
2
(8)
where, the average f 1 − score for the test samples
in the sequence length range ri is
¯
f 1
ri
, while f 1 is
the overall f 1 − score for the test dataset. A low
value indicated high consistency and vice versa.
3.2 Baseline Comparison Methods
A lot of work has been done in the past to predict
protein function using GO and amino acid sequences.
Notable works used for the fair comparison includes:
3.2.1 MLDA (Wang et al., 2016)
MLDA stands for Multi-Label Linear Discriminant
Analysis. This is based on the complete protein se-
quence that uses tf-idf features, further reduced in
dimension using the Multi-label LDA (MLDA) ap-
proach, as the input representation for the protein se-
quence. To reduce the features, MLDA project the
input feature to some other feature space.
3.2.2 ProtVecGen-Plus (Ranjan et al., 2019)
This work of ours, is the first to present the deep-
learning-based method that exploits the protein sub-
sequences to infer functional annotation(s) for the
full-length protein sequence. To infer protein func-
tions, multiple LSTM-based network architectures
are used, each trained with different-sized protein
subsequences (i.e., 100, 120, 140).
3.2.3 ProtVecGen-Ensemble (Ranjan et al.,
2021)
Another sub-sequence-based method, this time em-
ploying the tf-idf + MLDA technique. However, this
also entails discarding a few protein sub-sequences
that have been found to be less informative and re-
lying on the remaining sub-sequences to infer annota-
tion(s) for the full-length protein sequence.
3.2.4 ProtVecGen-Plus + MLDA (Ranjan et al.,
2019)
This is an ensemble of standard machine learning
and deep learning methods. The results of the
MLDA approach are combined with the results of
deep learning-based ProtVecGen-Plus (Ranjan et al.,
2019) .This method showed great potential for pre-
dicting protein functions.
A Sequence-Motif Based Approach to Protein Function Prediction via Deep-CNN Architecture
247

Table 3: Biological Process Dataset: Classification report with respect to different number of sub-networks with the proposed
approach on protein sub-sequences (N stands for number of sub-networks).
Dataset —> Full-length Sequence Approach Sub-Sequence Approach
S.
No.
N # parame-
ters (Mil-
lions)
Pr
avg
(%)
Rec
avg
(%)
F1
avg
(%)
CF Pr
avg
(%)
Rec
avg
(%)
F1
avg
(%)
CF
1. 6 ≈ 1.79 92.75 33.53 34.44 8.101 56.25 55.20 52.97 5.229
2. 7 ≈ 2.21 93.17 32.44 33.37 8.707 57.31 56.61 54.19 4.690
3. 8 ≈ 2.66 92.30 33.84 34.73 8.960 58.98 57.17 55.45 4.917
4. 9 ≈ 3.15 91.99 35.48 36.33 9.232 59.70 57.32 55.89 4.471
Table 4: Molecular Function Dataset: Classification report with respect to different number of sub-networks with the proposed
approach on protein sub-sequences (N stands for number of sub-networks).
Dataset —> Full-length Sequence Approach Sub-Sequence Approach
S.
No.
N # parame-
ters (Mil-
lions)
Pr
avg
(%)
Rec
avg
(%)
F1
avg
(%)
CF Pr
avg
(%)
Rec
avg
(%)
F1
avg
(%)
CF
1. 6 ≈ 1.67 94.34 45.40 s
46.24
5.722 71.23 70.94 69.09 5.013
2. 7 ≈ 2.07 93.24 50.82 51.35 5.544 71.42 71.14 69.34 4.970
3. 8 ≈ 2.50 94.35 49.58 50.35 5.938 72.25 72.02 70.08 5.125
4. 9 ≈ 2.97 93.68 49.84 50.84 5.481 72.72 72.21 70.57 4.057
3.3 Study the Effect of Number of
sub-Networks
In this sub-section, the effect of number of sub-
networks (denoted as N), considering N as 6, 7, 8,
9, with the proposed architecture is studied for both
the datasets. The experiments are conducted for two
different cases:
1. Full-length Protein Sequence: The proposed ar-
chitecture is trained and evaluated based on the
full-length protein sequences. The truncation of
protein sequences larger than 500 amino acids are
done.
2. Protein Sub-sequence: This represents the com-
plete proposed framework that is based on the pro-
tein sub-sequences.
This sort of study will allow to understand advan-
tages with the sub-sequence based method over the
methods that are based on the full-length sequence
model. The results for both cases are reported in Ta-
ble 3 (for the BP dataset) and Table 4 (for the MF
dataset).
On increasing the number of sub-networks, with
respect to various performance metrics, including
Pr
avg
, Re
avg
, and F1
avg
, a general increase is ob-
served. Further, increasing the number of sub-
networks also helps improve the consistency f actor,
a lower value of consistency f actor indicate a better
generalized behavior of the model towards protein se-
quences of different lengths. For the sub-sequence
based framework, the best F1
avg
reported are 55.89%
(for the BP dataset) and 70.57% (for the MF dataset),
with the nine sub-networks. These experimental ob-
servations stand true for full-length sequences as well.
Importantly, the results as shown in the Table
3 and the Table 4, clearly indicate that the pro-
posed sub-sequence based framework is superior. The
performances obtained for the full-length sequence
model are notably worse when compared to the sub-
sequence based framework, and this observation ap-
plies regardless of choice of the sub-networks. Ever-
more, the full-length sequence model tends to favor
a particular size of protein sequence more as quan-
tified with the consistency f actor. With respect to
the best F1
avg
, in comparison to the full-length se-
quence model, the sub-sequence based framework is
able to produce an improvements of 19.56% for the
BP dataset and 19.73% for the MF dataset.
The poor performance of the full-length sequence
model can be attributed primarily to the model’s in-
ability to efficiently retain the useful information.
This is because, the useful information is masked by
the presence of too much not useful information, es-
pecially for the case involving long-sized protein se-
quences.
ICAART 2023 - 15th International Conference on Agents and Artificial Intelligence
248
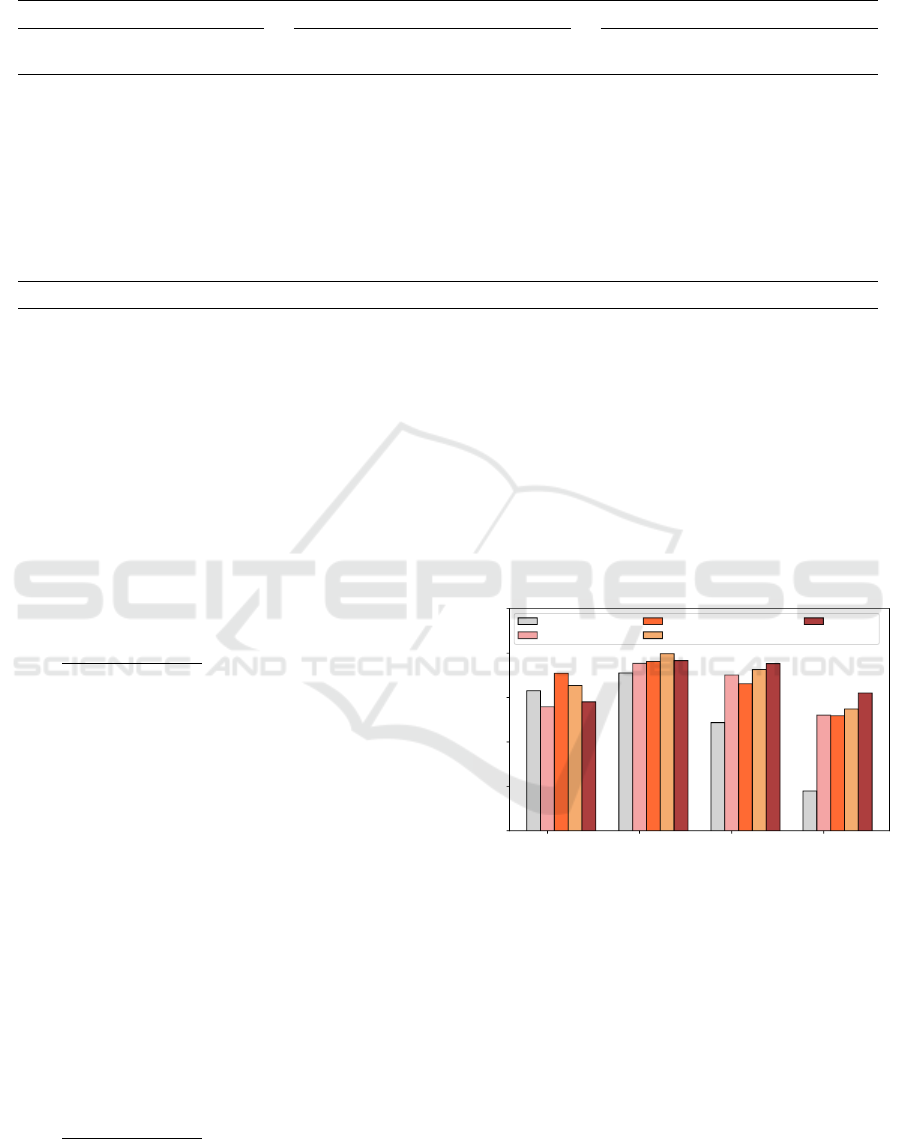
Table 5: Comparison between the state-of-art approach and proposed model (CF = consistency factor).
Dataset —> Biological Process Molecular Function
S.
No.
Approach Pr
avg
(%)
Rec
avg
(%)
F1
avg
(%)
CF Pr
avg
(%)
Rec
avg
(%)
F1
avg
(%)
CF
1. MLDA (Wang et al.,
2016)
52.61 49.42 49.27 10.969 60.20 58.29 57.91 8.408
2. ProtVecGen-Plus (Ran-
jan et al., 2019)
56.65 56.42 54.65 5.681 67.42 66.93 65.91 4.732
3. ProtVecGen-Ensemble
(Ranjan et al., 2021)
58.59 56.09 55.34 5.056 67.69 66.32 65.47 3.279
4. ProtVecGen-Plus +
MLDA (Ranjan et al.,
2019)
58.80 58.19 56.68 5.281 68.27 68.62 67.12 5.022
5. Proposed model 59.70 57.32 55.89 4.471 72.72 72.21 70.57 4.057
3.4 Overall Comparison with
State-of-the-Art Approaches
In this section, the proposed model is compared with
the existing state-of-the-art literature works, that in-
clude: (i) Multi-label LDA (MLDA) (Wang et al.,
2016), (ii) ProtVecGen-Plus (Ranjan et al., 2019),
(iii) ProtVecGen-Ensemble (Ranjan et al., 2021), and
(iv) hybrid approach ProtVec-Plus + MLDA (Ranjan
et al., 2019). The observed performance metrics for
each of the methods are shown in Table 5 for both the
BP and MF datasets.
3.4.1 [object Promise]
For the BP dataset, the proposed approach easily
betters the results with the MLDA (Wang et al.,
2016), ProtVecGen-Plus (Ranjan et al., 2019) and
ProtVecGen-Ensemble (Ranjan et al., 2021) ap-
proachs, the respective absolute enhancement in
F1
avg
being +6.62%, +1.24% and +0.55%, as shown
in Table 5. A similar trend is seen for the MF dataset
as well, with the proposed approach showing an im-
provement of +12.66%, +4.66%, and +5.10% over the
MLDA (Wang et al., 2016), ProtVecGen-Plus (Ranjan
et al., 2019), ProtVecGen-Ensemble (Ranjan et al.,
2021), respectively. The other metrics, Pr
avg
and
Re
avg
follow this behavior as well.
In comparison to the ProtVecGen-Plus + MLDA
(Ranjan et al., 2019), the proposed methods stood
second for the BP dataset, while comfortably outper-
forming for the MF dataset. The increase in the F1
avg
for the MF dataset is +3.45%.
3.4.2 [object Promise]
Hereby, the consistency f actor gives an indication
about the model’s behavior to perform for protein
sequences of various lengths. For the BP dataset,
the consistency f actor of the proposed model is re-
duced by 6.49, 1.21, 0.58, and 0.81 units with re-
spect to MLDA (Wang et al., 2016), ProtVecGen-Plus
(Ranjan et al., 2019), ProtVecGen-Ensemble (Ranjan
et al., 2021) and ProtVecGen-Plus + MLDA (Ran-
jan et al., 2019), respectively. For MF, the proposed
model stood next to the ProtVecGen-Ensemble (Ran-
jan et al., 2021) (consistency f actor = 3.279), while
reducing the consistency f actor by 4.35, 0.67, and
0.96 units over the MLDA, ProtVecGen-Plus, and
ProtVecGen-Plus + MLDA respectively.
0-200
201-500
501-1000
>1000
Protein Sequence Length Range ---->
20
30
40
50
60
70
Avg. F1-Scores ---->
MLDA
ProtVecGen-Plus
ProtVecGen-Ensemble
ProtVecGen-Plus+MLDA
Proposed
Figure 3: Biological Process: Length-wise performances of
protein sequences.
An in-depth, detailed illustration of the perfor-
mances obtained with different methods for handling
protein sequences of various lengths is provided by
grouping the test protein sequences into four groups,
are shown in Figures 3 (BP) and 4 (MF). The pro-
posed method is showing great performances for sig-
nificantly large protein sequences (having sequence
length > 500 amino acids).
A Sequence-Motif Based Approach to Protein Function Prediction via Deep-CNN Architecture
249
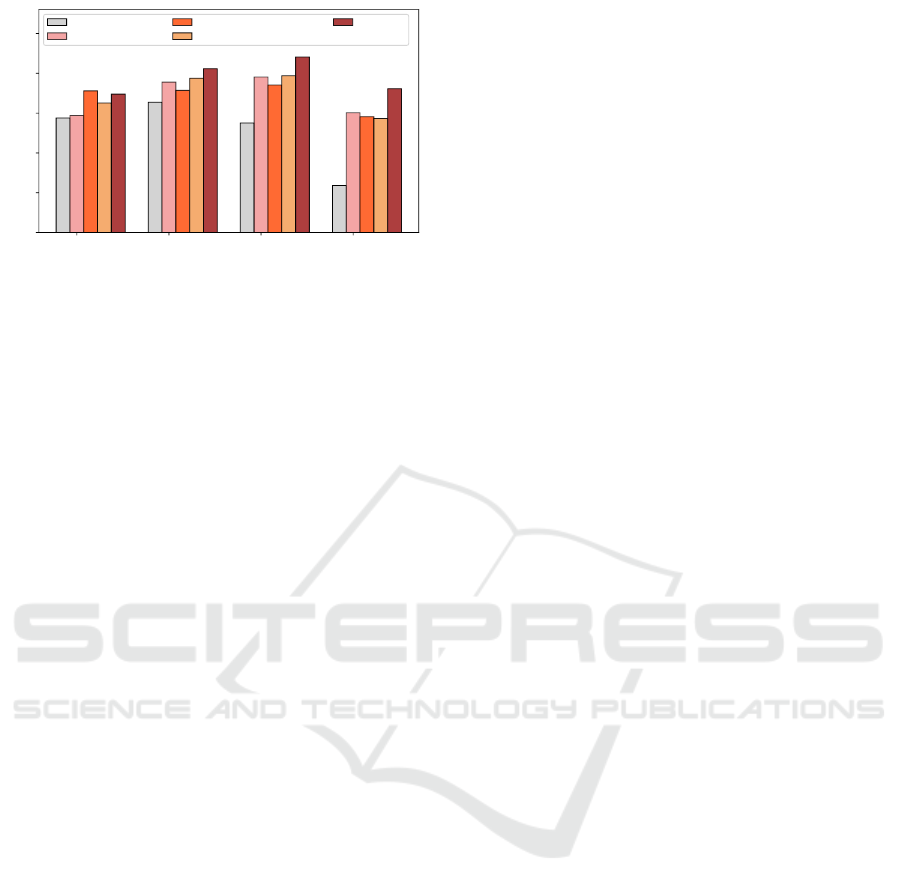
0-200
201-500
501-1000
>1000
Protein Sequence Length Range ---->
30
40
50
60
70
80
Avg. F1-Scores ---->
MLDA
ProtVecGen-Plus
ProtVecGen-Ensemble
ProtVecGen-Plus+MLDA
Proposed
Figure 4: Molecular Function: Length-wise performances
of protein sequences.
4 CONCLUSION
In this work, a sub-sequence based method for pro-
tein function prediction is introduced. The proposed
method takes benefits from information collected for
multiple sequence motifs – captured using the CNN
network – to determine the function for each sub-
sequence. Later, the functional inference for sub-
sequences are used to facilitate the functional annota-
tion of full-length protein sequence. Overall, the pro-
posed method showed great potential, especially for
long protein sequences. The research focused on pro-
tein sub-sequence is still an open research area, and
remarkably, can be great asset to improve the protein
studies. Future work will focus on merging additional
features and putting different deep learning models to
the test.
REFERENCES
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D.,
Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K.,
Dwight, S. S., Eppig, J. T., et al. (2000). Gene ontol-
ogy: tool for the unification of biology. Nature genet-
ics, 25(1):25–29.
Cao, R., Freitas, C., Chan, L., Sun, M., Jiang, H., and Chen,
Z. (2017). Prolango: protein function prediction using
neural machine translation based on a recurrent neural
network. Molecules, 22(10):1732.
Consortium, U. (2015). Uniprot: a hub for protein informa-
tion. Nucleic acids research, 43(D1):D204–D212.
Fa, R., Cozzetto, D., Wan, C., and Jones, D. T. (2018). Pre-
dicting human protein function with multi-task deep
neural networks. PloS one, 13(6):e0198216.
Gligorijevi
´
c, V., Renfrew, P. D., Kosciolek, T., Leman,
J. K., Berenberg, D., Vatanen, T., Chandler, C.,
Taylor, B. C., Fisk, I. M., Vlamakis, H., et al.
(2021). Structure-based protein function prediction
using graph convolutional networks. Nature commu-
nications, 12(1):1–14.
Ioffe, S. and Szegedy, C. (2015). Batch normalization: Ac-
celerating deep network training by reducing internal
covariate shift. In International conference on ma-
chine learning, pages 448–456. PMLR.
Jiang, Y., Oron, T. R., Clark, W. T., Bankapur, A. R.,
D’Andrea, D., Lepore, R., Funk, C. S., Kahanda, I.,
Verspoor, K. M., Ben-Hur, A., et al. (2016). An
expanded evaluation of protein function prediction
methods shows an improvement in accuracy. Genome
biology, 17(1):1–19.
Kingma, D. P. and Ba, J. (2014). Adam: A
method for stochastic optimization. arXiv preprint
arXiv:1412.6980.
Kulmanov, M. and Hoehndorf, R. (2020). Deepgoplus:
improved protein function prediction from sequence.
Bioinformatics, 36(2):422–429.
Kulmanov, M., Khan, M. A., and Hoehndorf, R. (2018).
Deepgo: predicting protein functions from sequence
and interactions using a deep ontology-aware classi-
fier. Bioinformatics, 34(4):660–668.
Kumari, D., Ranjan, A., and Deepak, A. (2019). Pro-
tein function prediction: Combining statistical fea-
tures with deep learning. In Proceedings of 2nd In-
ternational Conference on Advanced Computing and
Software Engineering (ICACSE).
Maas, A. L., Hannun, A. Y., Ng, A. Y., et al. (2013). Rec-
tifier nonlinearities improve neural network acoustic
models. In Proc. icml, volume 30, page 3. Citeseer.
Makrodimitris, S., van Ham, R. C., and Reinders, M. J.
(2019). Improving protein function prediction using
protein sequence and go-term similarities. Bioinfor-
matics, 35(7):1116–1124.
¨
Ozt
¨
urk, H.,
¨
Ozg
¨
ur, A., and Ozkirimli, E. (2018). Deepdta:
deep drug–target binding affinity prediction. Bioinfor-
matics, 34(17):i821–i829.
¨
Ozt
¨
urk, H., Ozkirimli, E., and
¨
Ozg
¨
ur, A. (2019). Wid-
edta: prediction of drug-target binding affinity. arXiv
preprint arXiv:1902.04166.
Radivojac, P., Clark, W. T., Oron, T. R., Schnoes, A. M.,
Wittkop, T., Sokolov, A., Graim, K., Funk, C., Ver-
spoor, K., Ben-Hur, A., et al. (2013). A large-scale
evaluation of computational protein function predic-
tion. Nature methods, 10(3):221–227.
Ranjan, A., Fahad, M. S., Fern
´
andez-Baca, D., Deepak, A.,
and Tripathi, S. (2019). Deep robust framework for
protein function prediction using variable-length pro-
tein sequences. IEEE/ACM Transactions on Computa-
tional Biology and Bioinformatics, 17(5):1648–1659.
Ranjan, A., Fernandez-Baca, D., Tripathi, S., and Deepak,
A. (2021). An ensemble tf-idf based approach to pro-
tein function prediction via sequence segmentation.
IEEE/ACM Transactions on Computational Biology
and Bioinformatics.
Wang, H., Yan, L., Huang, H., and Ding, C. (2016).
From protein sequence to protein function via multi-
label linear discriminant analysis. IEEE/ACM trans-
actions on computational biology and bioinformatics,
14(3):503–513.
ICAART 2023 - 15th International Conference on Agents and Artificial Intelligence
250

Yang, J., Yan, R., Roy, A., Xu, D., Poisson, J., and Zhang,
Y. (2015). The i-tasser suite: protein structure and
function prediction. Nature methods, 12(1):7–8.
You, R., Zhang, Z., Xiong, Y., Sun, F., Mamitsuka, H., and
Zhu, S. (2018). Golabeler: improving sequence-based
large-scale protein function prediction by learning to
rank. Bioinformatics, 34(14):2465–2473.
A Sequence-Motif Based Approach to Protein Function Prediction via Deep-CNN Architecture
251
