
Heterologous Expression of Fluorescent Protein Gene in E. Coli
DH5α
Yilin Li
1,*
and Chenyu Yang
2
1
Biomedical Science Department, Southern University of Science and Technology, No. 1088, Xueyuan Avenue, Nanshan
District, Shenzhen, China
2
Queen Mary College, Nanchang University, the Medical College, Block C, No. 999, Xuefu Avenue, Honggutan New
District, Nanchang, China
Keywords:
Gene Editing, E. Coli, Plasmid, Fluorescent Protein.
Abstract: Gene editing has opened a new field of molecular genetics research and the door for humans to understand,
identify, isolate and modify genes to create new species. This project aims to produce heterologous fluorescent
protein in E. coli cells by gene editing. By PCR amplification, the fluorescent protein gene has been obtained
from the plasmid carrier; the target gene has been connected to the carrier, and finally transformed into the
clone vector. The results of this project show that the fluorescent protein expressed by E. coli can be obtained,
and the heterologous protein gene can be expressed in E. coli.
1 INTRODUCTION
Gene editing is an emerging technology that modifies
specific target genes in an organism’s genome. It can
efficiently carry out site-specific genome editing,
showing great potential in gene research, gene
therapy and genetic improvement. The rapid growth
of the human population increases the demand for
meat and dairy products speedily, which drives the
demand for crops, biofuels and livestock. It is an
urgent problem to consider how to improve the
effective yield of major crops The gene-editing
technology can significantly improve crop yield by
increasing plant resistance and improving crop
quality to meet human needs (Ahmar, 2020).
Genome editing to correct disease-causing
mutations is a promising way to treat human diseases.
Gene editing-based therapy has the potential to treat
more than 10,000 human single-gene diseases such as
sickle cell disease (Park, 2021), and benefit many
more complex diseases (Memi, 2018). Research
shows that gene editing has also achieved results in
treating cancer and AIDS.
As a simple and programmable nuclease-based
genome-editing tool, the clustered regularly spaced
short palindromic repeats (CRISPR)/CRISPR-
associated protein 9 (Cas9) system dramatically
improves the ability to make precise changes in the
human genome. In recent years, rapid advances in
CRISPR-based technologies have expanded their
reach and promoted CRISPR-based therapies in
preclinical trials (Cui, 2018). The number of gene
therapy drugs has increased rapidly, showing
significant therapeutic effects on some cancers,
genetic diseases and infectious diseases, especially
after the emergence of the CRISPR technology in
2013.
E. coli is an essential tool in gene-editing
technology. It can be transformed into a variety of
functions and uses of cell factories, to achieve
enzymes production, monoclonal antibodies, etc. It
can also be used to catalyze biological and chemical
reactions, synthesize complex compound molecules
difficult to be synthesized before, and accelerate the
creation and development of new drugs.
Researchers have isolated a variety of fluorescent
proteins from living organisms, using molecular
biology techniques to evolve mutants that cover
almost the entire fluorescence spectrum. The red
fluorescent protein mCherry and green fluorescent
protein GFP are widely used. They have good
characteristics in fluorescence intensity, light
stability, acid-base resistance, maturity rate and other
aspects. They can be used as fusion protein labels and
applied in multi-fluorescent labelling imaging
systems.
410
Li, Y. and Yang, C.
Heterologous Expression of Fluorescent Protein Gene in E. Coli DH5α.
DOI: 10.5220/0012025200003633
In Proceedings of the 4th International Conference on Biotechnology and Biomedicine (ICBB 2022), pages 410-418
ISBN: 978-989-758-637-8
Copyright
c
2023 by SCITEPRESS – Science and Technology Publications, Lda. Under CC license (CC BY-NC-ND 4.0)
This project is based on gene-editing technology.
In this project, the fluorescent protein gene is
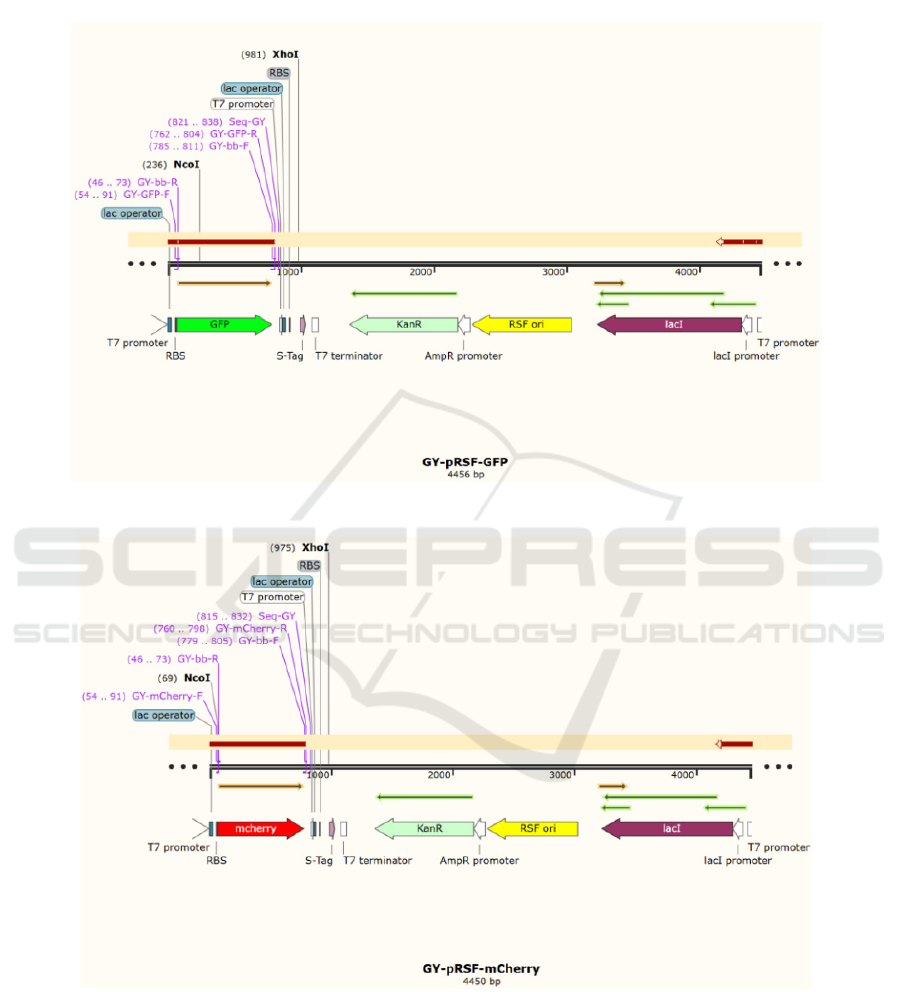
integrated onto prSFDUET-1 plasmid by the
homologous recombination technique, and the
recombinant plasmid is transformed into E. coli
BL21(DE3) cells. The fluorescent protein gene is
expressed in E.coli by induction, and the size of the
expressed protein is identified by SDS-PAGE.
2 MATERIALS AND METHODS
2.1 PCR Amplification of the Target
Gene
2.1.1 Reagents
Plasmids:
pRSFDuet-1, pET30a-mCherry, YEp181-TEF1-
GFP-FBA1.
Polymerases:
PrimeSTAR® Max DNA Polymerase enzyme,
Hieff® Taq DNA Polymerase.
Primers:
GY-mCherry-F: 5'-
TAATAAGGAGATATACCATGGTTTCCAAGG
GCGAGGAG-3';
GY-mCherry-R: 5'-
TCTGTTCGACTTAAGCATTACTTGTAGAGTT
CGTCCATG-3';
GY-GFP-F: 5'-
TAATAAGGAGATATACCATGCGTAAAGGAG
AAGAACTT-3';
GY-GFP-R: 5'-
TCTGTTCGACTTAAGCATTATTTGTATAGTTC
ATCCATGCCAT-3';
GY-bb-F: 5'-
TAATGCTTAAGTCGAACAGAAAGTAAT-3';
GY-bb-R: 5'-
CATGGTATATCTCCTTATTAAAGTTAAA-3'.
Others:
ddH
2
O, agarose, 1×TAE buffer, 5×loading
marker, DNA marker.
2.1.2 Materials
PCR tube (sterilized);
1.5 mL centrifuge tube (sterilized).
2.1.3 Apparatus
PCR machine, horizontal electrophoresis
2.1.4 Procedures
1) Miniprep plasmid (templates of PCR)
2) Single digestion of plasmid
3) Agarose gel detection of plasmid
4) PCR Reaction
Table 1: Reaction system: PCR Mixture in PCR tube.
Primestar polymerase 15 µL
Primer F 1.2 µL
Primer R 1.2 µL
plasmid template <10ng
ddH
2
O Up to 30 µL
total volume 30 µL
Calculate the number of volumes to be added for
pET30a-mCherry, pRSFDuet-1 and YEp181-TEF1-
GFP-FBA1 according to each reagent in the system
(ng/μL). Dosages are added to the PCR tubes in
descending order. Mix the reactants in a centrifuge
tube. Centrifugate for 5 s. Place the reaction tube into
the sample hole in the base of the PCR instrument, set
up the program, and conduct the PCR reaction:
Table 2: PCR Reaction Steps.
Preheat 94 ℃ 2 min
Denaturation 98 ℃ 10 s
Annealing 49 ℃ 5 s
Stretching 72 ℃ 40 s (10 s/kb)
Renaturation 72 ℃ 10 min
End 16 ℃ forever
Perform the denaturation, annealing and
extension cycles for 30 times. Then identify the
products of the PCR reaction.
The PCR products are identified whether
amplified or not by the pre-prepared 2% agarose gel
electrophoresis. Record the sequence of dot sampling
at 120 V and for 20 min as below.
Sampling: 4 μL PCR product + 1 μL 5× loading
buffer;
DNA marker: 5 mL;
2% agarose gel: 0.2 g agarose + 20 mL 1× TAE
buffer
The amplified product sizes: GFP (751 bp),
mCherry (711 bp), backbone (3745 bp).
The PCR products are purified and then stored in
a refrigerator at 4 ℃.
Heterologous Expression of Fluorescent Protein Gene in E. Coli DH5α
411

2.2 Double Fragment Homologous
Recombination
2.2.1 Recombination System Configuration
Carrier backbone=0.02×3745 bp=75 ng.
Fragment mCherry=0.04×711 bp=32 ng,
GFP=0.04×718 bp=32 ng
Table 3: ①pRSF-mCherry recombination.
System 20 μL
carrier backbone 75 ng
fragment mCherry 32 ng
5×CE II Buffer 4 µL
Exnase II 2 µL
ddH
2
O up to 20 µL
total volume 20 µL
Table 4: ②pRSF-GFP recombination.
System 20 μL
carrier backbone 75 ng
fragment GFP 32 ng
5×CE II Buffer 4 µL
Exnase II 2 µL
ddH
2
O up to 20 µL
total volume 20 µL
2.2.2 Recombination Reaction
The reaction takes place in a metal bath at 37 ℃ for
30 min, then the reagents are immediately placed on
ice for 5 min.
2.3 Transformation of the
Recombinant DNA Mixture into E.
Coli DH5Α Receptor Cells
2.3.1 Procedures
20 µL E. coli DH5α competent cells are removed
from a refrigerator at -80 ℃ and placed on an ice box.
Defrost at room temperature and are marked.
Add 5 µL of the recombinant system to the
competent cells, mix well, and take an ice bath for 30
min.
Recovery: operate heat shock for 90 s in a water
bath at 42 ℃, and then immediately place on the ice
box for 3 min.
Add 800 µL LB medium to each tube and culture
at 180 rpm, at 37 ℃, for 1 h.
Add 150 µL conversion solution to the
corresponding resistant (50 ng/µL kanamycin) plate
and evenly coat it with the liquid.
Place the plate upside-down in a constant-
temperature incubator at 37 ℃ for overnight
culturing.
2.4 Screening of Positive Clones by
Colony PCR
2.4.1 Materials
Conversion board, Hieff® Taq DNA Polymerase,
ddH
2
O, agarose, 1×TAE buffer, 5×loading marker,
DNA marker, PCR tubes (sterilized).
Primers:
GY-mCherry-F:5'-
TAATAAGGAGATATACCATGGTTTCCAAGG
GCGAGGAG-3'
GY-GFP-F:5'-
TAATAAGGAGATATACCATGCGTAAAGGAG
AAGAACTT-3'Carrier (0.5 μL)
Seq-GY(-R):5'-TTCGATTATGCGGCCGTG-3'
2.4.2 Procedures
Remove the plate transformed the night before from
the 37℃ incubator and draw a pat line.
Patch and amplify the bacterium colony.
Single colonies are enriched and cultured at 37 ℃
for 5-7 h after 20 µL ddH
2
O is added into the PCR
tube, and the bacteria coating is scraped into the PCR
tube at a ultra-clean table, with one tube for each
bacterium. After the thallus is fully dissolved in
ddH
2
O, the thallus DNA is released into the water by
lysis at 98 ℃ for 10 min. Centrifuge for 5 min, with
the supernatant used as the template DNA of the
colony PCR.
The following PCR systems are prepared.
Table 5: PCR Systems for Screening.
2×Hieff premix DNA
Polymerase
5 μL
Primer F 0.4 µL
Primer R 0.4 µL
Template 2 µL
ddH
2
O up to 10 µL
total volume 10 µL Mix
Mix and centrifuge the reactants for 5 s.
ICBB 2022 - International Conference on Biotechnology and Biomedicine
412

Place the reaction tube into the sample hole in the
base of the PCR instrument, set up the program, and
conduct the PCR reaction.
Table 6: PCR Reaction Steps.
Preheat 94 ℃ 5 min
Denaturation 94 ℃ 30 s
Annealing 58 ℃ 30 s
Stretching 72 ℃ 30 s (30 s/kb)
Renaturation 72 ℃ 10 min
End 16 ℃ forever
Perform the denaturation, annealing and
extension cycles for 30 times. The PCR products are
subjected to the pre-prepared 2% agarose gel
electrophoresis to identify whether the target products
are amplified. Record the sequence of the spot
sampling at 120 V and for 20 min.
Spot sample: 5 μL colony PCR product, DNA
marker 5 μL.
2% agarose gel: 0.2 g agarose + 20 mL 1× TAE
buffer.
The amplified product sizes: GFP positive
bacteria (785 bp), mCherry positive bacteria (779 bp).
2.5 Miniprep of Plasmid DNA
The colony PCR-verified positive strains are
inoculated into LB/K+ medium for overnight
culturing at 220 rpm at 37 ℃. Plasmid DNA is
extracted.
2.6 Enzyme Validation
The plasmid DNA is cleaved by a restriction
endonuclease.
2.6.1 Enzyme Digestion System
At 37 ℃ enzyme for 30 min.
Table 7: Enzyme digestion system.
system 10 μL
10×FastDigest buffer 1 µL
NcoI 0.2 µL
XhoI 0.2 µL
Template 500 ng
ddH
2
O Up to 10 µL
2.6.2 Identification of Enzyme Digestion
Results
The cleavage product bands of pRSF-GFP are
745+3711bp.
The cleavage product bands of pRSF-mCherry are
906+3544bp.
2.7 Plasmid DNA Transformed into E.
Coli BL21(DE3) Competent Cells
When the sequencing results are correct, the plasmid
DNA is transformed into E. coli BL21(DE3)
competent cells expressing the host.
2.8 mCherry/GFP Expression in E.
Coli BL21 (DE3) Induced by IPTG
2.8.1 Materials and Instruments
E. coli BL21 (DE3) containing pRSF-GFP plasmid,
E. coli BL21 (DE3) containing PRSF-mCherry
plasmid, LB/K+ solid and liquid medium, 1M IPTG
storage solution, kanamycin storage solution, 20mM
Tris-HCl buffer, 50ml, 2ml, 1.5ml centrifuge tubes,
pipette gun and head, hood spectrophotometer, low-
temperature centrifuge, ultra-clean table,
spectrophotometer.
2.8.2 Procedures
Strains containing recombinant plasmid are streaked
on LB plates containing kanamycin and cultured
overnight.
Single colonies are selected into about 5 mL LB
medium containing kanamycin and cultured
overnight at 37 ℃ and 220 rpm to obtain seed liquid.
The seeds are inoculated at 1:50 in 50 mL LB
medium containing kanamycin and cultured at 37 ℃
and 220 rpm for 1-1.5 h.
When OD600 is between 0.5 and 0.7, 25 µL IPTG
is added and induced at 180rpm, at 30 ℃ for 7 h.
Observe the colour of the solution.
After the induction, the bacterial solution OD is
measured, and the induced bacterial solution is
transferred to a 50 mL centrifuge tube. After being
centrifuged at 1340rpm for 30 min at low
temperature, the supernatant is discarded, and the
colour of the thallus is observed and photographed.
Resuspend with the buffer until 10OD/mL. Place 1 ml
of the sample on ice to be treated with an ultrasonic
crusher for 7 min (power 20%, 3 s each time. Interval
3 s).
Centrifuge at 12000 rpm for 10 min, transfer the
supernatant to another centrifuge tube, and precipitate
1 mL phase. Resuspend with the buffer. Add 80µL
supernatant or precipitate solution into the 1.5 mL
centrifuge tube, add 20µL 5× protein loading buffer
Heterologous Expression of Fluorescent Protein Gene in E. Coli DH5α
413
and mix evenly. Boil for 5 min at 100 ℃. Cool and
set aside at -20 ℃.
2.9 SDS-PAGE
2.9.1 Procedures
Preparation of protein gel.
Table 8. SDS-PAGE Gel formula
12% separation gel 5% stacking gel
Reagent
Volu
me
Reagent Volume
ddH
2
O
3.3
mL
ddH
2
O 2.2 mL
30%(w/v)
polyacrylam
ide
4 mL
30%(w/v)
polyacrylam
ide
0.67 mL
1.5 M Tris-
HCl(pH8.8)
2.5
mL
1.5 M Tris-
HCl(pH8.8)
1.0 mL
10% SDS
100
μL
10% SDS 40 μL
10% APS
100
μL
10% APS 40 μL
TEMED 6 μL TEMED 4 μL
Fix the gel glass plate on the electrophoresis
device and add trIS-glycine electrophoresis buffer in
the upper and lower tanks. Add the samples in
sequence, 10 µL solution for each aperture. Add an
equal volume of 1×SDS gel loading buffer to the
unused sample well.
Connect the electrophoresis device with the
power supply (positive electrode slot) and apply 90 V
on the gel. After the dye front enters the separation
glue, the voltage is increased to 120 V. The
electrophoresis continue until the bromophenol blue
reaches the bottom of the separation glue, and then
the power is turned off.
Remove the glass plate from the electrophoresis
device and carefully pry the glass plate.
Soak the gel in at least 5 times the volume of
dyeing solution and gently shake it on the
decolorizing shaker for at least 0.5 h.
Remove the dye. Soak the gel in the
decolorization solution, gently shake for 1-2 hours,
changing the decolorization solution 4 times.
3 RESULTS
3.1 Recombinant DNA
After the recombinant DNA is transformed into the
DH5α cells, the transformants on the plate are tested
by colony PCR.
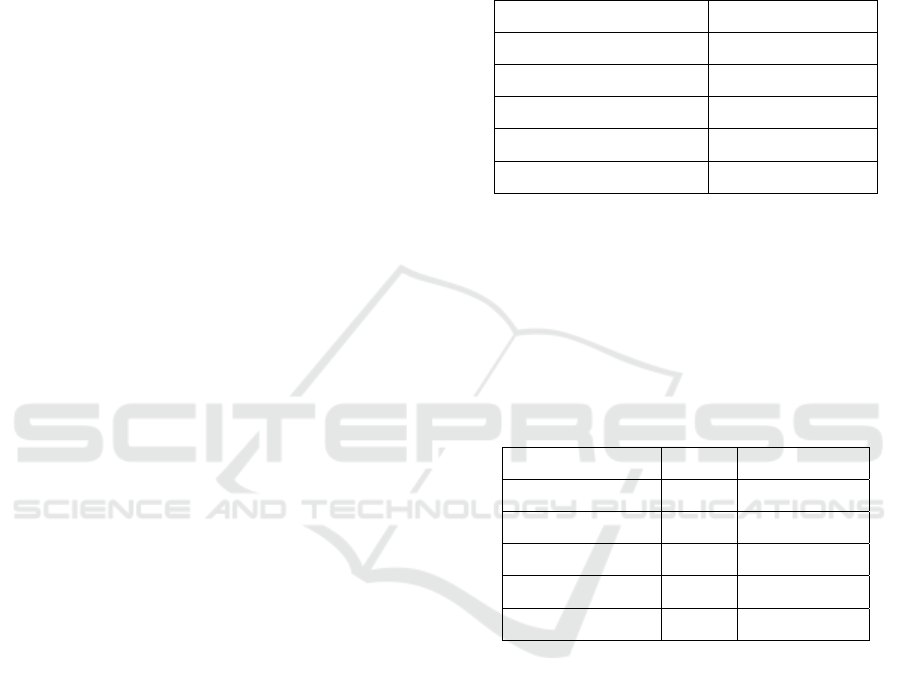
Figure 1: Colony PCR gel map.
Figure 1 shows that the colony is a positive colony
containing the target gene.
3.2 Plasmid Verification
To further verify whether the extracted plasmid DNA
is a positive clone, we cut the plasmid DNA using
restriction endonuclease to detect whether the target
band size is correct.
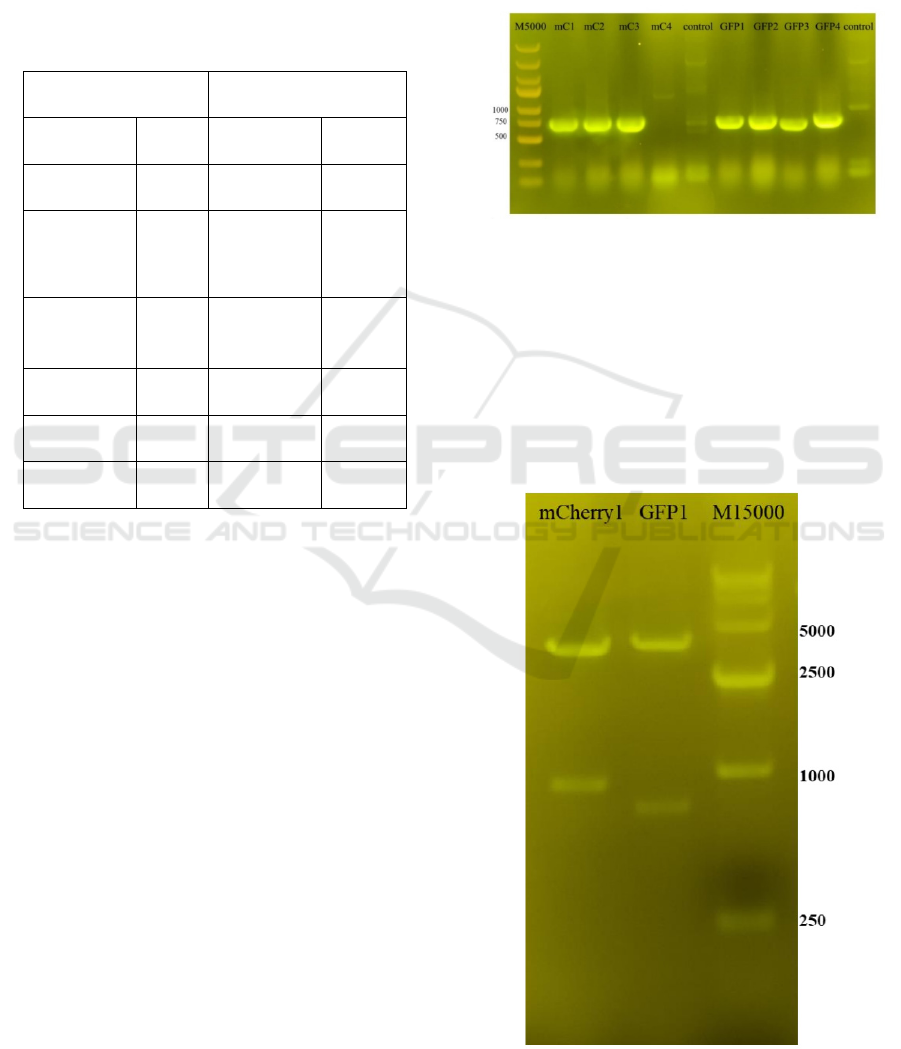
Figure 2: Enzyme digestion validation.
ICBB 2022 - International Conference on Biotechnology and Biomedicine
414
The results of enzyme digestion are correct
(Figure 2); the plasmid and primer (SEQ-Gy) are
sequenced and each identified by 4μL.
(a) dhwefhohc.
(b) fhrjfh.
Figure 3: Sequencing analysis.
Sequencing results show that mCherry is correct,
and there is a base mutation after the GFP starts codon
ATG. A) dhwefhohc. B) fhrjfh.
3.3 Fluorescence
IPTG is used as an inducer to induce the promoter
controlled by lactose operon to ensure stable
Heterologous Expression of Fluorescent Protein Gene in E. Coli DH5α
415

expression of heterologous protein in the condition of
good growth of E.coli and avoid the influence of a
large amount of heterologous protein expression on
the growth of E.coli.
(a) GFP and mCherry before induction (b) mCherry after induction (c) bacteria mCherry and GFP
Figure 4: Results of fluorescence.
Figure 5: Results of SDS-PAGE.
3.4 SDS-PAGE of mCherry and GFP
protein
The molecular weights of the products are verified by
SDS-PAGE protein polyacrylamide gel
electrophoresis.
ICBB 2022 - International Conference on Biotechnology and Biomedicine
416

4 CONCLUSION
This project demonstrates the basic operation model
of genetic engineering. The fluorescent protein gene
is integrated into the pRSFDuet-1 plasmid by
homologous recombination technology, and the
resulting recombinant plasmid is transformed into
E.coli BL21(DE3) cells, and the fluorescent protein
gene is expressed in E.coli by induction.
4.1 The Future of Gene Editing
The number of mutations in somatic cells increases
dramatically with age, some of which can lead to
cancer. In the future, gene-editing technology may
help modify specific regions of the genome to
prevent disease development.
In addition, the development of efficient genome-
editing tools, including base editors with high
specificity and no off-target effects, will open the
possibility of using them in human embryos to avoid
the spread of disease-causing mutations. Similarly,
newly developed CRISPR-free mitochondrial base-
editing methods are expected to correct pathogenic
mutations in mtDNA present in unfertilized oocytes
or embryos in the future and prevent transmission of
mitochondrial disease to the next generation (Li,
2020).
Genetic diseases are not only caused by mutations
in genes but also caused by low expression of the said
genes, thus affecting the function of tissues and
organs. During ageing, dysregulation of epigenetic
markers results in reduced expression of several
different genes that are important for the proper
functioning of cells and tissues, ultimately leading to
disease.
Since customizable nucleases have been shown to
regulate the expression of target genes without
modifying the genome sequence, they could be used
to treat a variety of diseases, improve cellular
characteristics of ageing, and restore tissue functions
(Reddy, 2020).
One of the major challenges in cancer
immunotherapy is targeting cancer cells specifically
while keeping healthy cells unharmed. Targeting
immune checkpoints through gene editing has been
shown to be a possible strategy (Ernst, 2020). In
treating primary tumors, targeted knockout can be
applied to viral infections such as HPV, which means
efficient gene knockout is required to reduce tumors
effectively. Targeting viral sequences rather than
endogenous genomic locations may be an advantage
to reduce the risk of unwanted genomic changes due
to gene editing. This could also be the potential
advantage of strategies to eradicate pre-HIV viruses
from the genome. HIV infection causes the body to
build up a dormant reservoir of proviruses, making it
much more difficult to cure (Wayengera, 2011). The
removal or destruction of HIV genes in vivo by
vector transport endonuclease can eliminate the
innate adaptive replication and survival of HIV and
provide an opportunity to prevent HIV gene
replication.
The way genome editing works presents a double-
edged sword: while it offers unprecedented
therapeutic benefits, it also has safety concerns.
Because a gene-editing therapy works by
producing DSBs in genomic DNA, the risk of
missing the target at an unintended site is higher than
that of another therapy that does not induce
chromosome insertion or genomic changes (Shim,
2017). Because therapeutic gene targeting strongly
relies on the creation of DSB at specific targets, in
vitro selection libraries (Guilinger, 2014), mismatch
detection nuclease assays (Vouillot, 2015), and
whole-genome sequencing (Gabriel, 2011) have been
developed to assess the targeting specificity of
nucleases. Studies have shown that base editors
(BEs) composed of a cytidine deaminase fused to
CRISPR/Cas9 enables efficient RNA-guided base
editing (Kim, 2017), which improve the efficiency of
nuclease editing. There is also the challenge of
developing efficient and safe methods to deliver
gene-editing elements to cells in the body. Existing
delivery methods, such as lipid nanoparticles
(Gilleron, 2013), are widely used. This method is safe
and simple but inefficient. In contrast, viral vectors
such as adenoviruses (Holkers, 2013) have higher
delivery efficiency, but there are unintentional
mutations and safety issues (Silva, 2011).
Moreover, unlike chemicals and antibody drugs,
gene-editing therapies require people to select
relevant animal models for safety studies. For in-vivo
gene-editing therapies, the binding specificity of the
designed nuclease is controlled by specific sequences
in the genome. Because the mouse genome is
substantially different from the human genome,
preclinical safety studies should instead be conducted
in animal models that mimic the human genome.
REFERENCES
Ahmar S, Saeed S, Khan MHU, Ullah Khan S, Mora-
Poblete F, Kamran M, Faheem A, Maqsood A, Rauf M,
Saleem S, Hong WJ, Jung KH. A Revolution toward
Gene-Editing Technology and Its Application to Crop
Improvement. Int J Mol Sci. 2020 Aug 7;21(16):5665.
Heterologous Expression of Fluorescent Protein Gene in E. Coli DH5α
417

doi: 10.3390/ijms21165665. PMID: 32784649;
PMCID: PMC7461041.
Cui Zhang, Renfu Quan, Jinfu Wang, Development and
application of CRISPR/Cas9 technologies in genomic
editing, Human Molecular Genetics, Volume 27, Issue
R2, 01 August 2018, Pages R79–R88,
https://doi.org/10.1093/hmg/ddy120.
Ernst, Martijn P T et al. “Ready for Repair? Gene Editing
Enters the Clinic for the Treatment of Human Disease.”
Molecular therapy. Methods & clinical development
vol. 18 532-557. 3 Jul. 2020,
doi:10.1016/j.omtm.2020.06.022.
Gabriel, R., Lombardo, A., Arens, A., Miller, J. C.,
Genovese, P., Kaeppel, C., Nowrouzi, A.,
Bartholomae, C. C., Wang, J., Friedman, G., Holmes,
M. C., Gregory, P. D., Glimm, H., Schmidt, M.,
Naldini, L., & von Kalle, C. (2011). An unbiased
genome-wide analysis of zinc-finger nuclease
specificity. Nature biotechnology, 29(9), 816–823.
https://doi.org/10.1038/nbt.1948.
Guilinger, J. P., Pattanayak, V., Reyon, D., Tsai, S. Q.,
Sander, J. D., Joung, J. K., & Liu, D. R. (2014). Broad
specificity profiling of TALENs results in engineered
nucleases with improved DNA-cleavage specificity.
Nature methods, 11(4), 429–435.
https://doi.org/10.1038/nmeth.2845.
Gilleron, J., Querbes, W., Zeigerer, A., Borodovsky, A.,
Marsico, G., Schubert, U., Manygoats, K., Seifert, S.,
Andree, C., Stöter, M., Epstein-Barash, H., Zhang, L.,
Koteliansky, V., Fitzgerald, K., Fava, E., Bickle, M.,
Kalaidzidis, Y., Akinc, A., Maier, M., & Zerial, M.
(2013). Image-based analysis of lipid nanoparticle-
mediated siRNA delivery, intracellular trafficking and
endosomal escape. Nature biotechnology, 31(7), 638–
646. https://doi.org/10.1038/nbt.2612.
Holkers, M., Maggio, I., Liu, J., Janssen, J. M., Miselli, F.,
Mussolino, C., Recchia, A., Cathomen, T., &
Gonçalves, M. A. (2013). Differential integrity of
TALE nuclease genes following adenoviral and
lentiviral vector gene transfer into human cells. Nucleic
acids research, 41(5), e63.
https://doi.org/10.1093/nar/gks1446.
Kim, K., Ryu, S. M., Kim, S. T., Baek, G., Kim, D., Lim,
K., Chung, E., Kim, S., & Kim, J. S. (2017). Highly
efficient RNA-guided base editing in mouse embryos.
Nature biotechnology, 35(5), 435–437.
https://doi.org/10.1038/nbt.3816.
Li, H., Yang, Y., Hong, W., Huang, M., Wu, M., & Zhao, X.
(2020). Applications of genome editing technology in
the targeted therapy of human diseases: mechanisms,
advances and prospects. Signal transduction and
targeted therapy, 5(1), 1.
https://doi.org/10.1038/s41392-019-0089-y.
Memi F, Ntokou A, Papangeli I. CRISPR/Cas9 gene-
editing: Research technologies, clinical applications
and ethical considerations. Semin Perinatol. 2018
Dec;42(8):487-500. doi:
10.1053/j.semperi.2018.09.003. Epub 2018 Oct 2.
PMID: 30482590.
Park SH, Bao G. CRISPR/Cas9 gene editing for curing
sickle cell disease. Transfus Apher Sci. 2021
Feb;60(1):103060. doi: 10.1016/j.transci.2021.103060.
Epub 2021 Jan 10. PMID: 33455878; PMCID:
PMC8049447.
Reddy P, Vilella F, Izpisua Belmonte JC, Simón C. Use
of Customizable Nucleases for Gene Editing and
Other Novel Applications. Genes (Basel). 2020 Aug
22;11(9):976. doi: 10.3390/genes11090976. PMID:
32842577; PMCID: PMC7565838.
Shim, Gayong et al. “Therapeutic gene editing: delivery
and regulatory perspectives.” Acta pharmacologica
Sinica vol. 38,6 (2017): 738-753.
doi:10.1038/aps.2017.2.
Silva, G., Poirot, L., Galetto, R., Smith, J., Montoya, G.,
Duchateau, P., & Pâques, F. (2011). Meganucleases and
other tools for targeted genome engineering:
perspectives and challenges for gene therapy. Current
gene therapy, 11(1), 11–27.
https://doi.org/10.2174/156652311794520111.
Vouillot, L., Thélie, A., & Pollet, N. (2015). Comparison of
T7E1 and surveyor mismatch cleavage assays to detect
mutations triggered by engineered nucleases. G3
(Bethesda, Md.), 5(3), 407–415.
https://doi.org/10.1534/g3.114.015834.
Wayengera, Misaki. “Proviral HIV-genome-wide and pol-
gene specific zinc finger nucleases: usability for
targeted HIV gene therapy.” Theoretical biology &
medical modelling vol. 8 26. 22 Jul. 2011,
doi:10.1186/1742-4682-8-26.
ICBB 2022 - International Conference on Biotechnology and Biomedicine
418
