
Efficient k-mer Indexing with Application to Mapping-free SNP
Genotyping
Mattia Marcolin, Francesco Andreace and Matteo Comin
Department of Information Engineering, University of Padua, Via Gradenigo 6/a, 35100 Padua, Italy
Keywords:
Genotyping, Mapping-free, k-mers Set Representation.
Abstract:
Advances in sequencing technologies and computational methods have enabled rapid and accurate identifica-
tion of genetic variants. Accurate genotype calls and allele frequency estimations are crucial for population
genomics analyses. One of the most demanding step in the genotyping pipeline is mapping reads to the human
reference genome. Recently mapping-free methods, like Lava and VarGeno, have been proposed for the geno-
typing problem. They are reported to perform 30 times faster than a standard alignment-based genotyping
pipeline while achieving comparable accuracy. Moreover, these methods are able to include known genomic
variants in the reference making read mapping, and genotyping variant-aware. However, in order to run they
require a large k-mers database, of about 60GB, to be loaded in memory. In this paper we study the prob-
lem of genotyping using new efficient data structures based on k-mers set compression, and we present a fast
mapping-free genotyping tool, named GenoLight. GenoLight reports accuracy results similar to the standard
pipeline, but it is up to 8 times faster. Also, GenoLight uses between 5 to 10 times less memory than the other
mapping-free tools, and it can be run on a laptop. Availability: https://github.com/CominLab/GenoLight.
1 INTRODUCTION
The discovery and characterization of sequence varia-
tions in human populations is crucial in genetic stud-
ies. A prime challenge is to efficiently analyze the
variations of a freshly sequenced individual with re-
spect to a reference genome and the available ge-
nomic variation data. Single nucleotide polymor-
phism (SNP) genotyping has been widely used in hu-
man disease-related research such as genome-wide
association studies (Stranger et al., 2011) and a recent
study on rare-disease diagnosis (NEJ, 2021).
The approaches to SNP genotyping can be
roughly divided into three categories: microarray
methods, sequencing alignment-based methods, and
alignment-free methods. The first approach uses SNP
arrays (Pastinen and et al., 2000). SNP arrays are fast
and inexpensive; however, they can only hold a lim-
ited number of probes: the state-of-the-art Affymetrix
genome-wide SNP array 6.0 has only 906 000 SNP
probes, compared to 31 million known common SNPs
in dbSNP (build 150).
The second approach is based on high-throughput
whole-genome sequencing and read alignment. In
most NGS-based genotyping pipelines, the first step
after sequencing a genome is to map each read to
the reference (Li and Durbin, 2010; Langmead and
Salzberg, 2012). Standard tools for genotyping (e.g.
Samtools mpileup (Li et al., 2009) and GATK Hap-
lotypeCaller (McKenna and et al., 2010)) require
this mapping information for every read before being
able to call variants. Yet, despite recent advances in
speed (Marco-Sola et al., 2012; Siragusa et al., 2013;
Yorukoglu et al., 2016; Zaharia et al., 2011), map-
ping still remains a computationally expensive step.
Furthermore, genotyping pipelines also include vari-
ant calling steps, significantly increasing the total run-
time.
The third approach is based on high-throughput
whole genome sequencing followed by an alignment-
free sequence comparison (Zielezinski et al., 2017).
Alignment-free methods have been used to save com-
pute time and memory by avoiding the cost of full-
scale alignment (Vinga, 2014). The reliance on a
single reference human genome could introduce sub-
stantial biases in downstream analyses (Brandt et al.,
2015; G
¨
unther and Nettelblad, 2019; Salavati et al.,
2019). Furthermore, including known variants in
the reference makes read mapping, variant calling,
and genotyping variant-aware. Alignment-free ap-
proaches have been applied to SNP genotyping (Sha-
jii et al., 2016; Sun and Medvedev, 2019). They
62
Marcolin, M., Andreace, F. and Comin, M.
Efficient k-mer Indexing with Application to Mapping-free SNP Genotyping.
DOI: 10.5220/0010985700003123
In Proceedings of the 15th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2022) - Volume 3: BIOINFORMATICS, pages 62-70
ISBN: 978-989-758-552-4; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

introduced two SNP genotyping tools named LAVA
and VarGeno, respectively, which build an index from
known SNPs (e.g., dbSNP) and then use approxi-
mate k-mer matching to genotype the donor from se-
quencing data. LAVA and VarGeno have been re-
ported to perform 4 to 30 times faster than a standard
alignment-based genotyping pipeline while achieving
comparable accuracy. Recently, another alignment-
free genotyping tool, called MALVA (Denti et al.,
2019), has been able to handle indels. Remark-
ably, alignment-free methods provide in some cases
even better results than the most widely adopted
SNPs discovery pipeline (Shajii et al., 2016; Sun
and Medvedev, 2019). In addition, these tools pro-
vide a faster alternative to read mapping, and with
the increasing of sequencing capabilities, they are a
promising direction of investigation. However, they
require a large amount of memory for indexing k-
mers, about 60GB, and all tools can be run only on
a large server with adequate RAM. In this work, we
introduce GenoLight that will address this problem
with new efficient data structures based on k-mer set
compression.
2 PRELIMINARIES
In this section, we report the basic concepts about the
functionality of VarGeno (Sun and Medvedev, 2019)
and the notation used inside this document to explain
GenoLight more clearly. We describe VarGeno be-
cause it is the fastest tool and also because the other
methods follow a similar paradigm. Given a database
of biallelic SNPs l
S
encoded as VCF (Variant Call-
ing Format) file, a database of reads from the genome
that we want to genotype, and a reference genome
G
R
, VarGeno produces a VCF file containing the most
probable genotype for each variant inside l
S
. SNPs
not contained in l
S
are not genotyped.
The VarGeno pipeline consists of two phases.
During the first phase, VarGeno maps each read r in-
side the database on G
R
, in an alignment-free fashion,
and then in the genotyping phase it uses the align-
ment information to detect the SNPs. To understand
how the mapping is performed, we need to introduce
some definitions and concepts. We denote by G
S
the
genome obtained by G
R
replacing, at each locus con-
taining an SNP ∈ l
S
, the alternative variant.
Conceptually, in this phase VarGeno extracts all
non-overlapping k-mer, with k= 32, from a read r and
searches for them in G
R
and G
S
. The position sup-
ported by a k-mer is the difference between the po-
sition in which the k-mer occurs inside the genome
(G
R
and/or G
S
) and the position in which it appears
inside the read. The position supported by the largest
number of k-mers and that respect two constraints that
we will define later in this section is called the target
position (t
p
) and represents the initial position where
the read is mapped to G
R
or G
S
. We report a simple
example in Figure 1, in which the read r is split into
three non-overlapping k-mers, that are searched into
the two genomes. All three k-mers are in agreement
and support the target position 10.
To search k-mers in G
R
and G
S
efficiently,
VarGeno builds two dictionaries D
re f
and D
snp
using
Bloom filers. D
re f
stores the binary encoding of each
overlapping k-mer, with k = 32, from G
R
and the rel-
ative initials positions. K-mers that occur inside G
R
with a frequency greater than 10 are discarded be-
cause they likely lead to an incorrect calculation of
t
p
.
The same process could be used to build D
snp
from G
S
. However, the only k-mers not present in
D
re f
would be the 32 consecutive k-mers for each
SNP ∈ l
S
that contain the alternative variant. So,
VarGeno stores only such k-mers inside D
snp
in a bi-
nary encoding with the relative initials positions.
Using a specific search algorithm that involves the
use of D
re f
and D
snp
, VarGeno is able to efficiently
obtain all the initial positions of a k-mer in G
R
and
G
S
and thus establishes the target position of a read.
The presence of sequencing errors inside the reads
can lead to an incorrect calculation of t
p
. To solve
this problem, given a k-mer K, LAVA (Shajii et al.,
2016) calculates the initial position within G
re f
and
G
snp
not only by searching for K but also of all k-mers
belonging to his Hamming neighbourhood, denoted
by N(K). This set is composed of all k-mers having,
at most, a Hamming distance equal to 1 from K. We
observe that |N(k)| = 3k + 1. In this way, k-mers that
would be present in G
R
or in G
S
, but are affected by a
single sequencing error contribute to the correct cal-
culation of the target position. To reduce the search
space and thus increase the temporal performance,
VarGeno uses quality scores to choose which k-mer
∈ N(K) to search within D
re f
and D
snp
(besides ob-
viously K). In particular, it searches for the k-mers
K
n
∈ N(K) whose character K
n
[i], 0 ≤ i ≤ 31 that dif-
ferentiates them from K has a quality score value Q[i]
higher than a certain threshold. We can now define
the constraint that the k-mer that supports position t
p
must satisfy. Let t
p
be the position supported by the
largest number of k-mers, to be the target position,
these two conditions must hold:
1. At least one of the k-mers extracted from r sup-
ports t
p
;
2. Let K
x
and K
y
be two k-mers extracted from r, x 6=
y. At least one k-mer belonging to the set N(K
x
)
Efficient k-mer Indexing with Application to Mapping-free SNP Genotyping
63

Figure 1: An example of target position calculation.
Figure 2: Pileup table update.
and one k-mer belonging to the set N(K
y
) must
support t
p
;
If more than one position has the same maxi-
mum number of supporter k-mers and respects these
two constraints, the current read is considered not
mapped. In general, if a read is not mapped, Vargeno
tries to map its reverse complement of r, performing
the same steps described.
The size of non-overlapping k-mers extracted
from reads, and consequently the size of k-mers inside
the dictionary, is fixed to 32 for the following reasons.
For the human genome, 85% of 32-mer have unit fre-
quency: this is fundamental for how t
p
is calculated.
The second reason is that the binary representation of
the k-mer can be stored exactly in a 64-bit variable
without wasting space in a 64-bit architecture.
Once the target position has been identified,
VarGeno checks if there are SNPs ∈ l
S
in the range
[t p, t p + |r|] within l
s
, where |r| is the length of the
read. If so, it proceeds with updating a data structure
called pileup table. For each SNP, there are two coun-
ters C
α
and C
β
. The first is increased by one unit if a
read contains the reference variant, and the second is
increased by one unit if the read contains the alterna-
tive variant. An example is shown in Figure 2.
Once all the dataset reads are processed, using the
data contained in the pileup table, VarGeno proceeds
with the genotyping phase, using the Bayesian statis-
tical framework (Sun and Medvedev, 2019).
3 METHODS
The basic idea of LAVA and VarGeno is to build two
dictionaries of k-mers, one with all k-mers from the
reference genome and another with the k-mers cov-
ering known SNPs. In LAVA, these dictionaries are
implemented with a hash table where all k-mers are
explicitly stored; instead, VarGeno uses a Bloom fil-
ter. Both these data structures need to be loaded into
memory for efficient queries, and they require about
60-63 GB of RAM. However, the size of these dic-
tionaries can be reduced because most of the infor-
mation carried by a k-mer is redundant. Given two
overlapping k-mers, it is possible to reassemble them
into a single (k+1)-mer, thus reducing the storage re-
quirement by k-1 bases. In GenoLight we further
exploit this observation: a set of k-mers, associated
with known SNPs, is assembled into a string, or set
of strings, that contains all of them. This proce-
dure allows us to store the whole dictionary in linear
form, reducing the memory requirements from O(kn)
to O(n) without losing information.
This problem is closely related to the represen-
tation and compression of k-mer sets, which has at-
tracted the attention of many researchers recently
(B
ˇ
rinda et al., 2021; Rahman and Medvedev, 2020).
K-mer sets are widely used in many bioinformatics
applications (Storato and Comin, 2021; Qian et al.,
2018; Marchiori and Comin, 2017; Qian and Comin,
2019; Andreace et al., 2021a; Andreace et al., 2021b).
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
64

Figure 3: An overview of the genotyping pipeline of GenoLight.
In fact, a k-mers set can be represented by its De
Bruijn graph. Instead of explicitly storing the graph, it
can be compressed by assembling the graph and com-
puting a string that traverses all nodes in the graph
only once. However, this problem is far from triv-
ial, like traditional assembly, and the algorithms pro-
posed in (B
ˇ
rinda et al., 2021; Rahman and Medvedev,
2020) will not produce a single string that traverses all
nodes. Instead, the results of this assembly are a set of
strings, called unitigs, that describe and cover the De
Bruijn graph. These techniques have been success-
fully applied to quality value compression (Shibuya
and Comin, 2019a; Shibuya and Comin, 2019b).
In this work, we use these memory efficient data
structures to calculate the target position of a read.
The aim of this project is to develop a tool that can
be run on a personal computer without the need of a
server with a large amount of RAM. Moreover, we
implement an efficient search algorithm with time
performance similar to that of other alignment-free
methods.
In order to use the efficient representation of k-
mer set (B
ˇ
rinda et al., 2021), the dictionary D
snp
needs to be replaced by two new data structures, the
RI reassembly index and a new dictionary named
D
SmartSnp
. Recall that D
snp
contains all k-mers ex-
tracted from G
S
that cover an SNP. The binary en-
coding of these 32-mer isn’t stored directly inside
D
SmartSnp
. Instead, these k-mers are compressed into
a string called the reassembly index, indicated with
RI, in which each k-mer extracted from G
S
is present
with unit frequency. In order to create the reassem-
bly index RI we use the Prophasm k-mer set assem-
bler (B
ˇ
rinda et al., 2021). Given a dataset of k-mers,
Prophasm creates a string that contains all input k-
mers exactly once. However, this compressed repre-
sentation has two characteristics that we need to con-
sider. First, depending on the input k-mers set, it may
not be possible to assemble all of them into a single
string. Instead, Prophasm produces a set of strings
that assemble and cover all input k-mers. The sec-
ond issue is that a k-mer can be represented in the
reassembly index in the forward or reverse comple-
ment orientation. Unfortunately, the orientation can-
not be determined a priori. Moreover, if in the input
k-mer set two k-mers that are one the reverse com-
plement of the other, these two k-mers will be repre-
sented only once in the reassembly index and not as
two separated k-mers. In summary, a set of k-mers
can be compressed using Prophasm into a reassem-
bly index, with a substantial saving in terms of space.
However, some important information might be lost
during this process, and for this reason we introduce
a new dictionary named D
SmartSnp
.
In GenoLight, to compute the target position of
a k-mer and the associated SNP position, we need to
store for each k-mer in the reassembly index: the orig-
inal position in the reference; its direction, forward
or reverse; and the associated SNP. Instead of storing
the k-mers in binary format as in D
snp
, every k-mer
in RI can be identified by its position in the reassem-
bly index. The new dictionary D
SmartSnp
contains a
set of records, one for each position in the reassembly
index RI. In a record, the most important informa-
tion is the original position of the k-mer inside G
S
.
While the binary encoding of a 32-length k-mer re-
quires 64 bits to be stored, the integer representing
the k-mer’s position can be stored using 32 bits. This
reduces the size of D
SmartSnp
compared to D
snp
by 32
bits for each record. The second information in the
record is the SNP position with respect to the k-mer.
To correctly calculate the target position, it is essential
to keep track of the k-mer orientation inside RI with
respect to G
S
. This information is also stored in the
k-mer record inside D
SmartSnp
. If the k-mer appears
both as a forward and reverse complement in G
s
, then
in the reassembly index it will appear only once, in
one of the two directions. In this case, the information
on the k-mer that is present inside the RI is stored in
D
SmartSnp
, while the information on other k-mer will
Efficient k-mer Indexing with Application to Mapping-free SNP Genotyping
65

be stored in a small auxiliary data structure.
The read mapping phase of GenoLight is simi-
lar to that of VarGeno. A read is split into non-
overlapping k-mers, and these k-mers are searched in
the reference sequence G
R
and the alternative refer-
ence G
S
, in order to detect a candidate target position,
see Figure 3. However, the representation of these
references is different with respect to VarGeno. In
VarGeno, both G
R
and G
S
are represented by Bloom
filters. Whereas in GenoLight, we use the original ref-
erence sequence for G
R
, and the reassembly index RI
and D
SmartSnp
for G
S
. Both G
R
and RI contain a set of
sequences in which we want to search for k-mers.
In order to allow for fast queries we use a vari-
ant of the FM-index (Ferragina and Manzini, 2000),
the FDM-Index also used in BWA (Li and Durbin,
2010). GenoLight uses FDM-Index to index G
R
and
RI, and the bound backtracking algorithm described
in (Li and Durbin, 2010) to search for k-mers in-
side it. This algorithm solves the inexact string match
problem and is used by the Burrows-Wheeler Aligner
(BWA). BWA performs local alignment of reads on a
genome according to the seed-and-extend alignment
paradigm. To perform the seeding phase efficiently, it
indexes the reference genome using FDM-Index and
performs searches with at most m mismatches to find
areas with high similarity using this algorithm. We
adapt this library allowing only for one mismatch and
using the additional constraint provided by quality
values so that possibly wrong bases are not aligned.
A pseudocode of the read mapping phase is re-
ported above. Each read is decomposed into non-
overlapping k-mers. For each k-mer we compute its
neighbourhood also using the quality values. Then,
these k-mers are searched in the reference genome
through its index. If a match is found, these positions
are stored in the vector P. To obtain the position p
G
S
where a k-mer k occurs inside G
S
, the alternative ref-
erence, GenoLight first identifies the starting position
p
reassembly
of k inside RI. Next, it accesses D
SmartSnp
using p
reassembly
as key to get p
G
S
. To efficiently
search k in RI, RI is also indexed using FDM-Index.
Once all matching positions have been stored in the
vector P, we can compute the target position of the
read, as in Figure 1. Finally, we can update the pileup
table and compute the genotyping using the Bayesian
statistical framework as in (Sun and Medvedev, 2019;
Shajii et al., 2016).
4 RESULTS
In this section, we report the results obtained by the
tools GenoLight, VarGeno (Sun and Medvedev, 2019)
Algorithm 1: GenoLight search.
Input:
IndexG: FDM-Index of the reference genome G
R
IndexRI: FDM-Index of the reassembly RI
D
SmartSnp
: Dictionary containing k-mer having the
alternative allele
r : Read to map into the reference genome
Q[K] : Quality scores associated with a k-mer K
extracted from r
Output
t
p
: target position of reads
Start
S ← ExtractNonOverlapKmer(r)
P ←
/
0
for each K ∈ S do
N(K)
f
← Filter(N(K), Q[K])
P ← SearchRe f erenceGenome(N(K)
f
, IndexG)
for each k ∈ N (K)
f
do
p
reassembly
← SearchRI(k, IndexRI)
p
Gs
← D
SmartSnp
[p
reassembly
]
P ← P
S
p
Gs
t
p
← CalculateTargetPosition(P)
End
and Lava (Shajii et al., 2016), as well as the standard
alignment-based genotyping pipeline (BWA (Li and
Durbin, 2010) as aligner, and BCFtools (Li, 2011)
as variant caller). The analysis focuses on two fun-
damental points, the computation time and memory
taken to complete the alignment and genotyping pro-
cess, and the accuracy of the results obtained by the
respective tools.
4.1 Experimental Setup
The datasets used in this study are two sets of real
reads from the same individual (NA12878) from the
1000 Genomes Project (Project, 2008): SRR622461
with a coverage of 6X (40 GB) and SRR622457 with
a coverage of 10X (65 GB). These datasets have
been widely used for benchmarking in other stud-
ies (Shibuya and Comin, 2019a; Shibuya and Comin,
2019b; Shajii et al., 2016; Sun and Medvedev, 2019;
Monsu and Comin, 2021). For validation, we used
an up-to-date high-quality genotype annotation gen-
erated by the Genome in a Bottle Consortium (Zook
et al., 2019). The GIAB gold standard contains
validated genotype information for NA12878, from
14 sequencing datasets with five sequencing tech-
nologies, seven read mappers, and three different
variant callers. The number of SNPs validated for
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
66

Table 1: Resources and Datasets used for testing.
DATASET DESCRIPTION
hg19 Reference genome
dbSNP Biallelic SNPs dataset (Sherry et al., 2001)
Affymetrix Affymetrix Genome-Wide Human SNP Array
NA12878 Gold standard from GIAB (Zook et al., 2019)
SRR622461 (Low Coverage) Dataset of reads with coverage 6X
SRR622457 (High Coverage) Dataset of reads with coverage 10X
NA12878 in the gold standard is 3.7M. The reference
genome used is hg19, also described as Genome Ref-
erence Consortium Human Build 37 (GRCh37). For
the SNPs dataset, we choose dbSNP (Sherry et al.,
2001), which contains human single nucleotide vari-
ations, and more specifically 11M SNPs biallelic,
and Affymetrix Genome-Wide Human SNP Array 6.0
with about 1M SNPs. A summary of the datasets used
for the evaluation is reported in Table 1.
To measure the accuracy, we use RTG Tools and
the SNP list which are also genotyped in the GIAB
gold standard. All tests were performed on a 14 lame
blade cluster DELL PowerEdge M600 where each
lame is equipped with two Intel Xeon E5450 at 3.00
GHz, 16GB RAM and two 72GB hard disk in RAID-
1 (mirroring).
4.2 Computational Resources: Time
and Memory
In this section we test the requirement of computa-
tional resources for all tools. Table 2 reports a sum-
mary of the results obtained for the two read datasets,
with the two SNP databases, for all tools.
We consider the standard alignment-based geno-
typing pipeline as the reference to compare the three
mapping-free algorithms. As expected, the standard
pipeline is always more time consuming while re-
quiring only 4 GB of memory. On the low coverage
dataset the standard pipeline requires 1929 minutes
to align 179 millions reads, that is more than 1 day
of computation. On this dataset VarGeno is able to
genotype the reads in only 59 and 44 minutes, de-
pending on the SNP databases, remaining the fastest
tool, thanks to the Bloom filter, as expected (Sun and
Medvedev, 2019). GenoLight is the second fastest
tool, it can process the low coverage dataset in 406
and 295 minutes, using the two SNP databases, which
results in a speed-up of 4.75x and 6.5x with respect to
the standard pipeline. LAVA is the slowest of the three
mapping-free methods.
If we consider the high coverage dataset, that con-
tains 287 million reads, the standard pipeline requires
2560 minutes, again more than one day of compu-
tation on a server. The time performance of the
mapping-free tools is similar to that of the low cov-
erage dataset. VarGeno is the fastest tool with 80 and
55 minutes, GenoLight needs 503 and 296 minutes,
and Lava requires 723 and 435 minutes, on the two
SNP databases. On the high coverage datasets, the
speed-up of GenoLight with respect to the standard
pipeline is 5.1x and 8.6x, for the two SNP databases.
GenoLight is again the second fastest tool; however,
on the high coverage dataset the speed-up w.r.t. stan-
dard pipeline increases.
In terms of memory requirement, the standard
pipeline is the least expensive, with only 4 GB of
RAM needed. For the mapping-free tools, the mem-
ory is mainly dominated by the size of the k-mers
database and the corresponding data structures, and
it varies only slightly with the number of the input
reads. On the smallest SNP database, e.g. Affymetrix,
GenoLight requires only 6 GB of RAM, whereas Lava
requires 57.6 GB and VarGeno about 59 GB. On the
largest SNP database, e.g. dbSNP, GenoLight needs
12.5 GB of memory, whereas Lava requires 60 GB
and VarGeno 63 GB.
In summary, only GenoLight and the standard
pipeline can run on a laptop with 16 GB of RAM
because the memory requirements of both methods
are low. However, GenoLight is much faster than the
standard pipeline, up to 8.6x.
4.3 Genotyping Accuracy
In this section we test the genotyping accuracy of all
tools on both datasets. The VCF file produced by the
standard pipeline and all mapping-free tools are com-
pared with the gold standard (Zook et al., 2019). In
Table 3 are shown the results in terms of accuracy, in
line with other studies (Shajii et al., 2016; Sun and
Medvedev, 2019).
For the Low Coverage dataset, the standard
pipeline has an accuracy of 93%. VarGeno and Geno-
Light have similar performance, varying from 91% to
93.5%, depending on the SNP databases. Lava shows
a lower accuracy in some cases. The different per-
formance of Lava can be explained by the fact that
it does not use quality value information, unlike the
Efficient k-mer Indexing with Application to Mapping-free SNP Genotyping
67
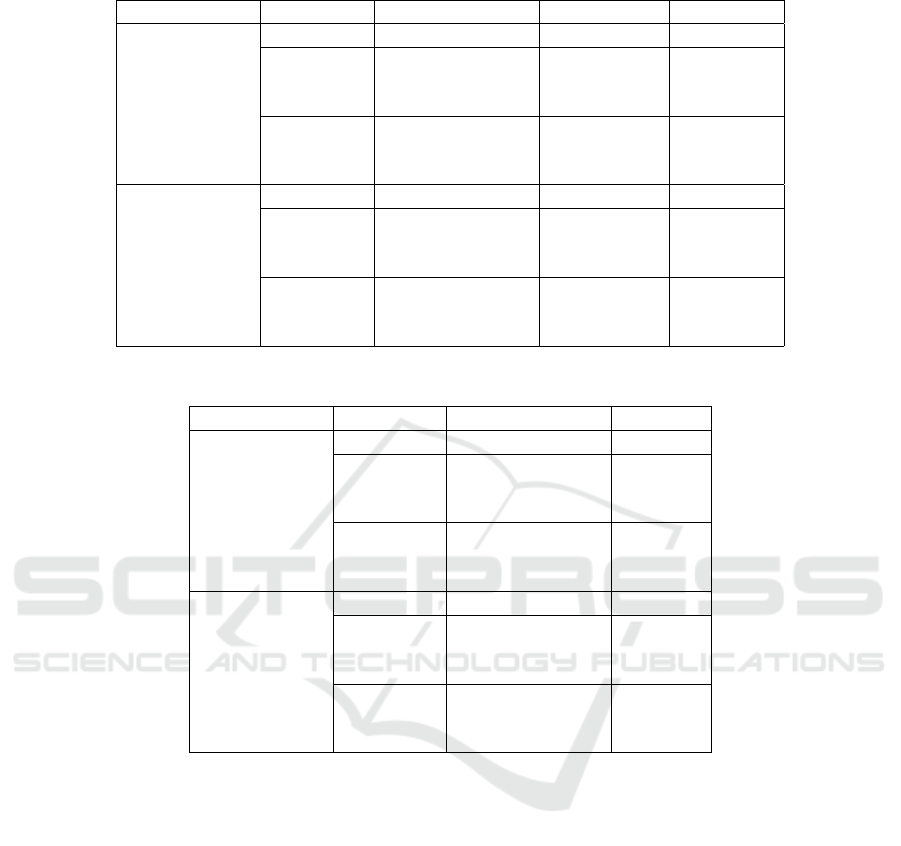
Table 2: Performance results of the genotyping for all tools on various datasets.
Dataset SNP db Algorithm Time (d:h:m) RAM (GB)
Low Coverage
None Standard Pipeline 1:08:09 4
dbSNP
VarGeno 59 63.2
Lava 8:26 61
GenoLight 6:46 12.5
Affymetrix
VarGeno 44 58.9
Lava 5:18 57.6
GenoLight 4:55 6
High Coverage
None Standard Pipeline 1:18:40 4
dbSNP
VarGeno 1:20 63.242
Lava 12:03 60
GenoLight 8:23 12.5
Affymetrix
VarGeno 55 59
Lava 7:15 57.6
GenoLight 4:56 6
Table 3: Performance results of the genotyping for all tools on various datasets.
Dataset SNP db Algorithm Accuracy
Low Coverage
None Standard Pipeline 0.930
dbSNP
VarGeno 0.911
Lava 0.819
GenoLight 0.912
Affymetrix
VarGeno 0.935
Lava 0.935
GenoLight 0.934
High Coverage
None Standard Pipeline 0.969
dbSNP
VarGeno 0.949
Lava 0.845
GenoLight 0.951
Affymetrix
VarGeno 0.977
Lava 0.974
GenoLight 0.976
other two mapping-free methods.
On the High Coverage dataset, thanks to the 10X
coverage, all tools reported higher accuracy values.
In this case, the standard pipeline has an accuracy
of 96.9%. Also, on this dataset the behaviour of all
mapping-free tools is similar to the previous case.
VarGeno and GenoLight have similar accuracies, in
line with the standard pipeline, whereas Lava is less
precise.
In summary, among the mapping-free tools,
VarGeno and Genolight achieve the best overall per-
formance in terms of accuracy, in line with the stan-
dard pipeline. GenoLight reports precision results
comparable to VarGeno, but it uses between 5 and 10
times less memory than the other two tools. The stan-
dard alignment-based pipeline is extremely slow, and
it requires more than a day of computation on a clus-
ter. GenoLight achieves almost the precision levels
and memory usage of the standard pipeline while be-
ing significantly faster, thus resolving the high mem-
ory issue of VarGeno and Lava.
5 CONCLUSIONS
In this paper we presented GenoLight an algorithm
to speed up the alignment-based genotyping of reads
with application to SNP detection. In the case of
SNP detection, a k-mer database can be exploited for
efficiently mapping of reads. Popular mapping-free
tools, like Lava and VarGeno, require a large amount
of memory to store these k-mers databases, more than
60 GB of RAM. In GenoLight we introduce a novel
k-mers set compression technique that allows to store
the same information in limited space, less than 12.5
GB. We tested different tools in popular benchmark-
ing datasets for SNP genotyping. The results show
that GenoLight is able to detect SNP with an accuracy
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
68

similar to that of the standard pipeline, in line with
VarGeno. However, GenoLight is up to 8 times faster
than the standard pipeline, while requiring a limited
amount of RAM, and it can be executed on a standard
laptop, unlike the other mapping-free tools. As a fu-
ture direction of investigation, it would be interesting
to extend GenoLight for the detection of other genetic
variations such as insertions and deletions.
REFERENCES
(2021). 100,000 genomes pilot on rare-disease diagnosis
in health care — preliminary report. New England
Journal of Medicine, 385(20):1868–1880.
Andreace, F., Pizzi, C., and Comin, M. (2021a). Metaprob
2: Improving unsupervised metagenomic binning
with efficient reads assembly using minimizers. In
Jha, S. K., M
˘
andoiu, I., Rajasekaran, S., Skums, P.,
and Zelikovsky, A., editors, Computational Advances
in Bio and Medical Sciences, pages 15–25, Cham.
Springer International Publishing.
Andreace, F., Pizzi, C., and Comin, M. (2021b). Metaprob
2: Metagenomic reads binning based on assembly
using minimizers and k-mers statistics. Journal of
Computational Biology, 28(11):1052–1062. PMID:
34448593.
B
ˇ
rinda, K., Baym, M. H., and Kucherov, G. (2021). Sim-
plitigs as an efficient and scalable representation of de
bruijn graphs. Genome Biology, 22.
Brandt, D. Y. C., Aguiar, V. R. C., Bitarello, B. D., Nunes,
K., Goudet, J., and Meyer, D. (2015). Mapping
Bias Overestimates Reference Allele Frequencies at
the HLA Genes in the 1000 Genomes Project Phase I
Data. G3 Genes—Genomes—Genetics, 5(5):931–941.
Denti, L., Previtali, M., Bernardini, G., Sch
¨
onhuth, A.,
and Bonizzoni, P. (2019). Malva: Genotyping
by mapping-free allele detection of known variants.
iScience, 18:20 – 27.
Ferragina, P. and Manzini, G. (2000). Opportunistic data
structures with applications. In Proceedings 41st An-
nual Symposium on Foundations of Computer Sci-
ence, pages 390–398.
G
¨
unther, T. and Nettelblad, C. (2019). The presence and
impact of reference bias on population genomic stud-
ies of prehistoric human populations. PLOS Genetics,
15(7):1–20.
Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read
alignment with bowtie 2. Nature Methods, 9:357–359.
Li, H. (2011). A statistical framework for snp calling, mu-
tation discovery, association mapping and population
genetical parameter estimation from sequencing data.
Bioinformatics, 27 21:2987–93.
Li, H. and Durbin, R. (2010). Fast and accurate long-read
alignment with Burrows–Wheeler transform. Bioin-
formatics, 26(5):589–595.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J.,
Homer, N., Marth, G., Abecasis, G., and Durbin, R.
(2009). The sequence alignment/map format and sam-
tools. Bioinformatics, 25:2078–2079.
Marchiori, D. and Comin, M. (2017). Skraken: Fast and
sensitive classification of short metagenomic reads
based on filtering uninformative k-mers. In BIOIN-
FORMATICS 2017 - 8th International Conference
on Bioinformatics Models, Methods and Algorithms,
Proceedings; Part of 10th International Joint Confer-
ence on Biomedical Engineering Systems and Tech-
nologies, BIOSTEC 2017, volume 3, pages 59–67.
Marco-Sola, S., Sammeth, M., Guig
´
o, R., and Ribeca, P.
(2012). The gem mapper: fast, accurate and versa-
tile alignment by filtration. Nature Methods, 9:1185–
1188.
McKenna, A. and et al. (2010). The genome analysis
toolkit: a mapreduce framework for analyzing next-
generation dna sequencing data. Genome research,
20:1297–1303.
Monsu, M. and Comin, M. (2021). Fast alignment of
reads to a variation graph with application to snp de-
tection. Journal of Integrative Bioinformatics, page
20210032.
Pastinen, T. and et al. (2000). A system for spe-
cific, high-throughput genotyping by allele-specific
primer extension on microarrays. Genome research,
10(7):1031–42.
Project, . G. (2008). Igsr and the 1000 genomes project.
Qian, J. and Comin, M. (2019). Metacon: Unsupervised
clustering of metagenomic contigs with probabilistic
k-mers statistics and coverage. BMC Bioinformatics,
20(367).
Qian, J., Marchiori, D., and Comin, M. (2018). Fast and
sensitive classification of short metagenomic reads
with skraken. In Peixoto, N., Silveira, M., Ali, H. H.,
Maciel, C., and van den Broek, E. L., editors, Biomed-
ical Engineering Systems and Technologies, pages
212–226, Cham. Springer International Publishing.
Rahman, A. and Medvedev, P. (2020). Representation
of k-mer sets using spectrum-preserving string sets.
bioRxiv.
Salavati, M., Bush, S. J., Palma-Vera, S., McCulloch, M.
E. B., Hume, D. A., and Clark, E. L. (2019). Elimina-
tion of reference mapping bias reveals robust immune
related allele-specific expression in crossbred sheep.
Frontiers in Genetics, 10:863.
Shajii, A., Yorukoglu, D., Yu, Y. W., and Berger, B. (2016).
Fast genotyping of known snps through approximate
k-mer matching. Bioinformatics, 32:538–544.
Sherry, S. T., Ward, M.-H., Kholodov, M., Baker, J., Phan,
L., Smigielski, E. M., and Sirotkin, K. (2001). dbSNP:
the NCBI database of genetic variation. Nucleic Acids
Research, 29(1):308–311.
Shibuya, Y. and Comin, M. (2019a). Better quality score
compression through sequence-based quality smooth-
ing. BMC Bioinformatics, 20. (Impact Factor 2.9).
Shibuya, Y. and Comin, M. (2019b). Indexing k-mers in
linear space for quality value compression. Journal of
Bioinformatics and Computational Biology, 17(5).
Siragusa, E., Weese, D., and Reinert, K. (2013). Fast and ac-
curate read mapping with approximate seeds and mul-
Efficient k-mer Indexing with Application to Mapping-free SNP Genotyping
69

tiple backtracking. Nucleic Acids Research, 41:e78 –
e78.
Storato, D. and Comin, M. (2021). K2mem: Discover-
ing discriminative k-mers from sequencing data for
metagenomic reads classification. IEEE/ACM Trans-
actions on Computational Biology and Bioinformat-
ics, pages 1–1.
Stranger, B. E., Stahl, E. A., and Raj, T. (2011). Progress
and Promise of Genome-Wide Association Stud-
ies for Human Complex Trait Genetics. Genetics,
187(2):367–383.
Sun, C. and Medvedev, P. (2019). Toward fast and accurate
snp genotyping from whole genome sequencing data
for bedside diagnostics. Bioinformatics, 35:415–420.
Vinga, S. (2014). Alignment-free methods in computational
biology. Brief Bioinform., 15:341–342.
Yorukoglu, D., Yu, Y. W., Peng, J., and Berger, B. (2016).
Compressive mapping for next-generation sequenc-
ing. Nature Biotechnology, 34:374–376.
Zaharia, M. A., Bolosky, W. J., Curtis, K., Fox, A., Patter-
son, D. A., Shenker, S., Stoica, I., Karp, R. M., and
Sittler, T. (2011). Faster and more accurate sequence
alignment with snap. ArXiv, abs/1111.5572.
Zielezinski, A., Vinga, S., Almeida, J., and et al. (2017).
Alignment-free sequence comparison: benefits, appli-
cations, and tools. Genome Biol.
Zook, J., McDaniel, J., Olson, N., Wagner, J., Parikh, H.,
Heaton, H., Irvine, S., Trigg, L., Truty, R., McLean,
C., De La Vega, F., Xiao, C., Sherry, S., and Salit, M.
(2019). An open resource for accurately benchmark-
ing small variant and reference calls. Nature biotech-
nology, 37:561–566.
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
70
