
VAEResTL: A Novel Generative Model for Designing Complementarity
Determining Region of Antibody for SARS-CoV-2
Saeed Khalilian
2
, Zahra Moti
3
, Arian Baloochestani
4
, Yeganeh Hallaj
4
, Alireza Chavosh
1
and Zahra Hemmatian
1,∗
1
MarWell Bio Inc., California, U.S.A.
2
Independent Researcher, Iran
3
Independent Researcher, The Netherlands
4
Independent Researcher, Norway
Keywords:
Antibody, Nanobody, Complementarity Determining Region (CDR), SARS-CoV-2, COVID-19, Deep
Generative Models, Transfer Learning, Bioinformatics, in-silico Screening.
Abstract:
The global impact of the COVID-19 pandemic underlines the importance of developing a competent machine
learning (ML) approach that can rapidly design therapeutics and prophylactics such as antibodies/nanobodies
against novel viral infections despite data shortage problems and sequence complexity. Here, we propose
a novel end-to-end deep generative model based on convolutional Variational Autoencoder (VAE), Resid-
ual Neural Network (Resnet), and Transfer Learning (TL), named VAEResTL that can competently generate
CDR-H3 sequences for a novel target lacking sufficient training data. We further demonstrate that our pro-
posed method generates the third complementarity-determining region (CDR) of the heavy chain (CDR-H3)
sequences for designing and developing therapeutic antibodies/nanobodies that can bind to different variants
of SARS-CoV-2 despite the shortage of SARS-CoV-2 training data. The predicted CDR-H3 sequences are
then screened and filtered for their developability parameters namely viscosity, clearance, solubility, stability,
and immunogenicity through several in-silico steps resulting in a list of highly optimized lead candidates.
1 INTRODUCTION
Antibodies play an important role in therapeutic dis-
covery and vaccine development for a variety of dis-
eases ranging from infectious diseases to cancer and
autoimmune diseases (Zohar and Alter, 2020). Wet-
lab methods for antibody discovery can be very time-
consuming and costly. One of these methods is high
throughput screening which is a drug discovery pro-
cess used to identify the antibody leads that bind
to their antigen targets and are within the therapeu-
tics and developability index range (Sharma et al.,
2014). The binding site of antibody/nanobody in-
cludes a region known as complementarity determin-
ing region (CDR) (Murphy et al., 2008). Amongst
CDRs, CDR-H3 on the heavy chain is the most
variable CDR and typically contributes the most
to antigen specificity for antibodies and nanobod-
ies (Tsuchiya and Mizuguchi, 2016). The current
COVID-19 pandemic underlines the importance of
developing approaches capable of rapidly designing
and developing therapeutics and prophylactics against
∗
Corresponding author email: zara@marwell.bio
novel viral infections. Designing CDR-H3 plays
a critical role in antibody/nanobody-based therapeu-
tics. Artificial intelligence (AI) and machine learn-
ing (ML) techniques have been recently explored for
COVID-19 vaccine development to stop the spread of
the virus (Ong et al., 2020); however, rapid develop-
ment of antibodies and nanobodies which can offer
therapeutics and prophylactics benefits is of critical
importance.
Recently, biologically plausible deep learn-
ing (Yoo et al., 2020), and computational mod-
els (Adolf-Bryfogle et al., 2018) have been success-
fully applied to design and optimize CDR loops us-
ing deep sequencing data (Norman et al., 2020).
Deep generative models using Variational Autoen-
coder (VAE) have also been applied in designing pro-
teins (Friedensohn et al., 2020), and in predicting
protein structures (Guo et al., 2020). Moreover, deep
Residual Neural Network models (Resnet) have also
greatly improved protein design and protein structure
predictions (Lu et al., 2020), and antibody-epitope
classification (Ripoll et al., 2021). Nevertheless, the
application of Resnet in antibody/nanobody discov-
Khalilian, S., Moti, Z., Baloochestani, A., Hallaj, Y., Chavosh, A. and Hemmatian, Z.
VAEResTL: A Novel Generative Model for Designing Complementarity Determining Region of Antibody for SARS-CoV-2.
DOI: 10.5220/0010823700003123
In Proceedings of the 15th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2022) - Volume 3: BIOINFORMATICS, pages 107-114
ISBN: 978-989-758-552-4; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
107

ery has been rarely explored. Existing deep learning
and VAE-based approaches are often used in conjunc-
tion with large datasets and are incapable of discov-
ering and designing new antibodies/nanobodies when
facing data shortage for novel targets such as SARS-
CoV-2 and its variants. Transfer learning (TL) tech-
niques succeeded in biomedical image classification
and various protein prediction tasks (Heinzinger et al.,
2019; Valeri et al., 2020); however, to the best of
our knowledge, no study in the literature has empow-
ered the deep generative model of VAE with TL to
tackle the lack of training data in antibody/nanobody
discovery. Here, we leverage the power of deep
learning algorithms to predict therapeutic antibodies
with binding ability to SARS-CoV-2. We present a
novel end-to-end generative ”VAEResTL” model to
discover amino acid sequences against novel target
antigens that lack training data. Our proposed VAER-
esTL model is based on a VAE model, a Resnet struc-
ture, and a Network-based TL technique. We demon-
strate Resnet improved our deep generative model
VAERes’s performance efficiently while providing a
deep neural network capable of learning CDR-H3 se-
quence complexity. The learning from our VAERes
pre-trained on sufficient antibody amino acid CDR-
H3 sequences on different target antigens can be effi-
ciently transferred for predicting antibody/nanobody
amino acid sequences with binding ability to SARS-
CoV-2. The VAEResTL predicted CDR-H3 se-
quences were then subject to in-silico screening and
filtering for developability parameters namely viscos-
ity, clearance, solubility, stability, and immunogenic-
ity, resulting in potential lead antibody/nanobody
CDR-H3 sequence candidates with optimal therapeu-
tic properties.
2 METHODOLOGY
2.1 Datasets and Data Pre-processing
We used amino acid sequences of antibody CDR-H3
derived from deep sequencing data to develop and
train our proposed methods. We filtered out CDR-
H3 sequences based on their binding ability to SARS-
CoV-2 (Raybould et al., 2021) resulting in 2298 se-
quences which we used as our primary training data.
For our TL method, we used three large datasets of
three different antigenic targets with a wide variety
of antibodies, including ranibizumab (Rani) (the size
of the dataset is 67769 sequences) (Liu et al., 2020),
yeast display scFv (Yeast) (the size of the dataset is
11038 sequences) (Adams et al., 2016), and chicken
ovalbumin (OVA) (the size of the dataset is 65638 se-
quences) (Goldstein et al., 2019) to pre-train our al-
gorithms.
We converted amino acid sequences into a 2-
dimensional matrix through one-hot encoding. To
have the sequences of various lengths of 8-20 with the
fixed lengths of 20, we used padding and added null
character J to the left and right sides of sequences.
Since there are 24 amino acids (20 standards, two rare
(U, O), one unknown (X), and one null (J)), a CDR-
H3 sequence with a length of 20 amino acids results
in a 20 x 24 matrix. Each row will consist of a single
‘1’ in the column corresponding to the amino acid in
that position, whereby this value for all other columns
in that row is equal to ‘0’.
Figure 1: Overview of the proposed method.
2.2 Model Architecture and Training
The VAEResTL, which is an end-to-end trainable
model comprises a convolutional VAE that adopts a
Resnet structure and is enhanced with a TL technique
(Figure 1). Our method utilizes only the amino acid
sequences data without the need for structural data.
CDR-H3 sequences are subject to high variation in
the amino acid distribution and offer the highest con-
tribution to antigen specificity. A deep neural network
capable of learning from complex and highly variant
sequences is essential to model CDR-H3 sequences;
nevertheless, increasing the depth of the neural net-
work by adding more layers leads to a vanishing gra-
dient problem (Goceri, 2019). Resnet introduces skip
connections to jump over some layers. The skip con-
nections allow gradients of a deep neural network to
flow easily from layer to layer and prevent gradients
from vanishing (He et al., 2016). Inspired by the suc-
cessful applications of Resnet in bioinformatics (Xu
et al., 2020), (Ripoll et al., 2021), here we incorpo-
rate Resnet blocks into our convolutional VAE model
to increase the depth of the neural network for mod-
eling and predicting CDR-H3 sequences. We named
our convolutional VAE that adopts a Resnet approach,
VAERes. We further incorporated a network-based
TL technique (Tan et al., 2018), into our VAERes and
named our model VAEResTL (Figure 1).
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
108

3 EXPERIMENTS
3.1 Experimental Setup
We executed five experiments and compared VAE,
VAERes, VAEResTL with baseline models of HMM
and LSTM (Table 1). In experiment 1 and 2, we
trained the convolutional VAE and VAERes directly
on the SARS-CoV-2 dataset. For our VAEResTL we
designed experiment 3, through which VAERes was
trained on all the data together including Rani + Yeast
+ OVA sequences. Then the VAERes pre-trained on
Rani+ Yeast+ OVA sequences was trained on SARS-
CoV-2 dataset. In experiments 4 and 5, we trained the
baseline models of HMM and LSTM directly on the
SARS-CoV-2 dataset. HMM and LSTM are based on
sequence models described as follows:
(a) LSTM: We adopted an LSTM model previously
reported (Gupta et al., 2018), and replaced one-
hot encoding with embedding layer. The network
consists of two layers of LSTM with 100 units,
cross-entropy loss function and Adam optimizer.
(b) HMM: We used HMM model described by Ra-
biner (Rabiner, 1989) as a character-based model
where amino acid characters are considered as
states. Each state has a probability distribution
over a set of possible sequences. Amino acid
characters are then selected to form CDR-H3 se-
quences.
3.2 Experimental Metrics
We used machine learning (ML) and biophysical met-
rics to evaluate our proposed methods’ performance.
We further performed in-silico screening to assess
the predicted antibody/nanobody CDR-H3 sequences
that may bind to SARS-CoV-2.
3.2.1 Sequence Similarity
We used three different metrics to evaluate sequence
similarity. 1) Bilingual Evaluation Understudy
(BLEU) (Papineni et al., 2002), We employed 2-
gram, 3-gram, and 4-gram to estimate BLEU, using
the nltk python library. A higher BLEU score indi-
cates a higher degree of similarity between the seed
and the generated sequences. 2) Statistical measures
of Jensen-Shannon divergence (JSD) (Lin, 1991),
When the JSD value is close to zero, the distribution
of the generated sequences is very close to that of seed
sequences. 3) Pairwise sequence similarity method
of Needleman-Wunch (NW) (Needleman and Wun-
sch, 1970) is a biophysical characteristic and is used
to evaluate the sequence similarity between seed and
generated sequences (Wang et al., 2020), the higher
the NW value, the more similar the two sequences.
3.2.2 Sequence Diversity
We estimated the sequence diversity by measuring
the number of shared n-grams for different values
of n between generated and seed sequences, referred
to as S
n
(Das et al., 2018). Therefore, a value of
S
model1
n
/S
model2
n
< 1 implies more diversity of gener-
ated sequences by model 1 at a particular n compared
to that of model 2.
3.2.3 Biophysical Metrics
We used Bio and modLAMP library (M
¨
uller et al.,
2017) which incorporates several modules, like de-
scriptor calculation of biophysical characteristics
of amino acid CDR-H3 sequences, e.g., stability,
isoelectric-point, charge, and hydrophobicity (H) to
evaluate the biophysical properties of predicted CDR-
H3 sequences (Sharma et al., 2014).
3.2.4 In-Silico Screening
We used the CamSol method, a protein solubility pre-
dictor (Sormanni et al., 2015), at pH= 7.0 to esti-
mate the protein solubility score for each CDR-H3
sequence. We measured each sequence variant’s net-
charge and hydrophobicity (H) (Sharma et al., 2014)
to predict the sequences’ viscosity and clearance. We
predicted the peptide binding affinity of the vari-
ant CDR-H3 sequences to MHC Class II molecules
to a reference set of 26 human leukocyte antigen
(HLA) alleles by employing NetMHCIIpan (Jensen
et al., 2018) to reduce their immunogenicity. The
NetMHCIIpan’ output provides a percentile rank that
reflects sequences’ affinity compared with a set of
random natural peptides. The percentile rank classi-
fies the peptides weak and strong binders to specific
MHC Class II alleles. The strong binders have per-
centile rank of above two, and the weak binders have
percentile rank of below ten. The minimum percentile
rank, with percentile rank of below ten is also classi-
fied as weak binders, and the average percentile rank
is calculated across all 26 HLA alleles. The weaker
the peptide affinity binding, the less immunogenic is
a sequence (Mason et al., 2019).
VAEResTL: A Novel Generative Model for Designing Complementarity Determining Region of Antibody for SARS-CoV-2
109

Table 1: Baseline models comparison. Biological Characteristics and Machine Learning Metrics for generated SARS-CoV-2
CDR-H3 sequences by VAERes, VAEResTL, HMM, LSTM, and Seed SARS-CoV-2.
Method
Biophysical Characteristics Machine Learning Metrics
NW H ISO Charge Stability
BLEU
JSD
(2-gram) (3-gram) (4-gram)
VAERes 25.07 0.26 5.08 -2.15 13.18 76.56 75.66 75.59 0.007
VAEResTL 39.31 0.24 5.77 -1.20 25.46 77.57 76.41 75.20 0.005
LSTM 40.51 0.20 4.20 -1.60 57.48 81.12 80.01 78.54 0.11
HMM 9.12 0.52 9.5 0.68 45.9 82.1 80.50 78.60 0.009
Seed NA 0.23 5.64 -0.84 33.36 NA NA NA NA
4 EXPERIMENTAL RESULTS
AND DISCUSSION
4.1 VAE Models Comparison
The convolutional VAE algorithm could not learn
the complexity of CDR-H3 sequences, and 100%
of the generated amino acid sequences were abnor-
mal (invalid padding or invalid characters) and in-
appropriate for further analysis. Nevertheless, when
we incorporated Resnet structure into the convolu-
tional VAE, our VAERes model demonstrated signifi-
cant improvements in generating CDR-H3 sequences
for SARS-CoV-2. When we trained our VAERes
on SARS-CoV-2 seed CDR-H3 sequences, 98% of
the generated sequences were valid (valid padding
with valid characters), and only 2% of the gener-
ated sequences were abnormal (invalid padding or
invalid characters). Notwithstanding, 86.8% of the
valid generated CDR-H3 sequences for SARS-CoV-
2 were duplicate sequences, and only 11.2% of the
generated sequences were unique. These results sug-
gest that VAERes could learn a small range of CDR-
H3 sequences, reflecting the lack of adequate train-
ing data for SARS-CoV-2 seed CDR-H3 sequences
as a new target. With VAEResTL, 100% of the
generated CDR-H3 sequences for SARS-CoV-2 were
valid (valid padding with valid characters), 70.7% of
the valid generated sequences were unique, and only
29.3% of the CDR-H3 sequences were duplicate se-
quences.
4.2 Baseline Models Comparison
When we trained HMM on SARS-CoV-2 seed se-
quences, HMM could generate a very small library
of CDR-H3 sequences which is 1/56 of the size of
VAEResTL-generated library of SARS-CoV-2 CDR-
H3, where only 17% of the generated sequences
were valid (valid padding with valid characters) and
unique. These results may indicate that HMM as a
classic model is incapable of learning the complex-
ity of CDR-H3 sequences while suffering from lack
of sufficient training data. LSTM could also gen-
erate a small library of sequences, where 95.5% of
the sequences were valid (valid padding with valid
characters). However, 86.1% of the valid generated
CDR-H3 sequences for SARS-CoV-2 were duplicate
sequences, and only 9.4% of the valid sequences
were unique. LSTM could only generate a library of
SARS-CoV-2 CDR-H3 sequences which is 1/6 of
the size of VAEResTL-generated library of CDR-H3
sequences. We calculated the value for a number of
antibody heuristics including Needleman (NW), Hy-
drophobicity (H), Isoelectric Point (ISO), Charge, and
Stability that give biological clues about how VAERes
and VAEResTL perform compared to the baseline
models of HMM and LSTM (Table 1). The aver-
age pairwise sequence similarity of NW is consis-
tently lower for VAERes, and HMM than VAEResTL.
Though, the higher NW for LSTM can be due to the
bias of such a small ratio of the unique sequences
among the small library of valid sequences. The av-
erage H value for VAEResTL generated sequences is
closer to the seed sequences than generated sequences
by VAERes, HMM, and LSTM. The ISO values are
between 4.20 and 5.77 across all the other models that
are close to the ISO values for the seed sequences,
except the HMM generated sequences that has the
highest ISO value. The average Charge values for all
models are negative except for HMM. The average
stability values for VAERes generated sequences are
too low. The average stability values for LSTM gen-
erated sequences and HMM generated sequences are
too high. Therefore, predicted sequences by VAERes,
LSTM, and HMM are not appropriate for their bio-
physical stability property. Moreover, the VAER-
esTL has a stability value within the range of seed
sequences. The overall BLEU values for VAEResTL-
generated sequences have higher values for 2-gram,
3-gram, and 4-gram than the BLEU for VAERes. The
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
110
small ratio of valid and unique sequences for LSTM
and HMM may bias their learning ability to only
a small range of CDR-H3 sequences, reflecting the
higher BLEU values for LSTM and HMM. Nonethe-
less, the JSD value for LSTM and HMM is higher
than other models presented in Table 1. The over-
all ML and Biophysical metrics demonstrate VAER-
esTL can more efficiently predict CDR-H3 sequences
with binding ability to SARS-CoV-2. It is likely that
with transfer learning, VAEResTL learns a more “bi-
ologically plausible” latent space by utilizing a much
larger dataset than VARes and the baseline models
of LSTM and HMM. These results may further in-
dicate that VAEResTL architecture loads more bio-
logical context during the CDR-H3 sequence gener-
ation process. Our baseline model comparison anal-
ysis may suggest that VAEResTL outperforms base-
line model techniques by predicting a more exten-
sive library of valid and biologically more valuable
antibody/nanobody CDR-H3 sequences with binding
ability to SARS-CoV-2 more accurately despite the
shortage of training data.
4.3 Transfer Learning Impact
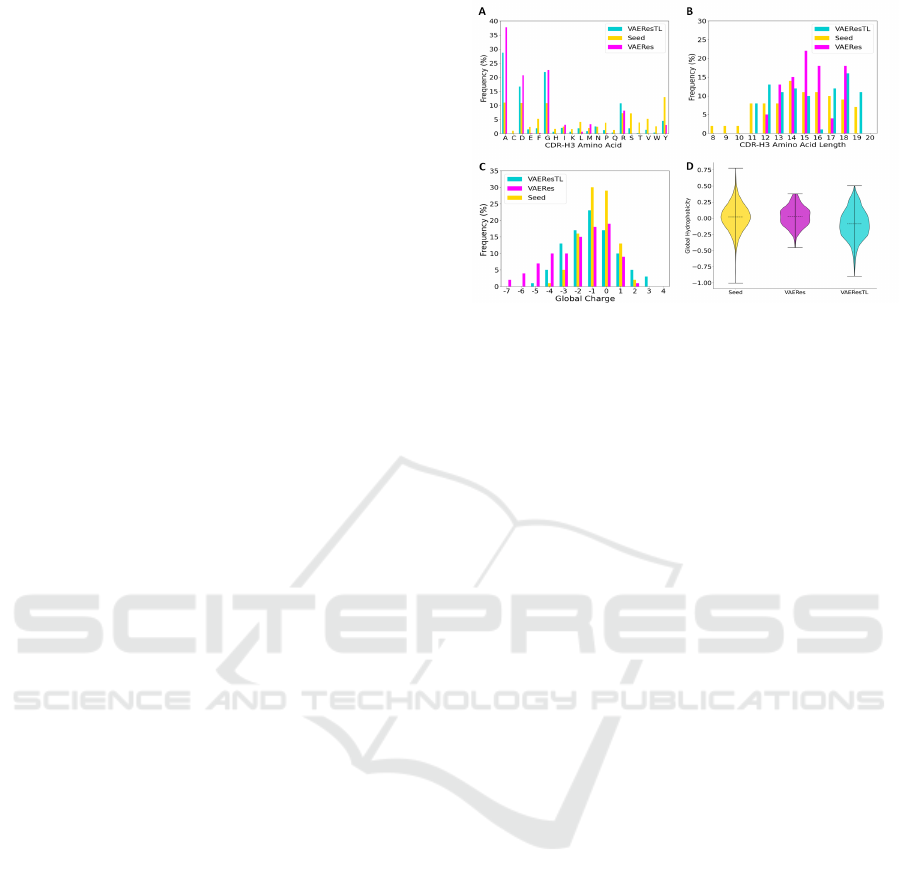
Figure 2 compares predicted sequences by VAERes,
VAEResTL, with the seed CDR-H3 sequences that
bind to SARS-CoV-2 to visually imply the impact of
TL. We reported molecular features, e.g., sequence
length, amino acid sequence distribution, charge, and
H, as they play a crucial role in determining the
membrane-binding affinity and specificity. We report
the VAEResTL results when our proposed method
is pre-trained by Rani + Yeast + OVA. The aver-
age amino acid composition-frequency distribution
(Figure 2A), length distribution (Figure 2B), net-
charge (Figure 2C), and H (Figure 2D) of VAEResTL-
generated SARS-CoV-2 CDR-H3 match the seed se-
quences more than the VAERes-generated SARS-
CoV-2 CDR-H3. This observation may suggest that
VAERes with TL perform well in capturing the charge
patterning, H, and composition within generated se-
quences.
We further analyzed the diversity of the gener-
ated sequences in terms of their shared n-grams (S
n
).
The n-gram similarity is lower for VAEResTL with
respect to VAERes(S
VAEResT L
n
/S
VAERes
n
< 1) for n >
2. 2-gram, 3-gram, 4-gram, 5-gram are 0.91, 0.78,
0.63 and 0.44 respectively. These results imply that
VAEResTL-generated sequences show strong long-
range diversity; however, they are still consistent with
biological sequences, as evident from the sequence
similarity comparison (Table 1). The VAEResTL-
generated sequences show more substantial diversity
Figure 2: Visualize Comparison of molecular charac-
teristics between Seed (SARS-CoV-2 seed sequences, Yel-
low), VAERes (VAERes-generated sequences for SARS-
CoV-2, Violet) and VAEResTL (VAEResTL-generated se-
quences for SARS-CoV-2, Turquoise). Horizontal dashed
lines account for the mean. Whiskers extend to the most ex-
treme non-outlier data points. (A) amino acid distribution,
(B) amino acid length distribution, (C) total charge distribu-
tion, (D) Eisenberg hydrophobicity.
at higher n-grams (lower S
n
values) as a desirable
feature that can prevent viral resistance while de-
signing next-generation anti-virals and can provide a
larger and more diverse pool of sequences for in-silico
screening and therapeutics development studies. We
also found from the ML visualization heatmaps (Fig-
ure 3A1-3C1) and logo plots (Figure 3A2-3C2) that
although VAEResTL changed certain CDR-H3 amino
acid positions and their frequency distributions, the
overall pattern of VAEResTL-generated sequences
are closer to seed sequences as compared to the
VAERes-generated CDR-H3 sequences. High simi-
larity of predicted CDR-H3 sequences and seed CDR-
H3 sequences observed by biophysical and ML analy-
sis may suggest that the VAEResTL-generated CDR-
H3 sequences also have binding ability to SARS-
CoV-2. Moreover, ML and biophysical characteristics
of the VAEResTL-generated CDR-H3 sequences (Ta-
ble 1, Figure 2, and Figure 3) demonstrate that VAER-
esTL predicts more biologically valuable CDR-H3
sequences with binding ability to SARS-CoV-2 al-
though, we used divers training databases. These
results show that the generalization for our VAER-
esTL in antibody/nanobody design is significantly im-
proved.
4.4 In-Silico Screening
With current advances in computational fore-
casts (Raybould et al., 2019), a number of parame-
ters including viscosity, clearance, solubility, stabil-
ity, and immunogenicity are used as the guideline for
VAEResTL: A Novel Generative Model for Designing Complementarity Determining Region of Antibody for SARS-CoV-2
111
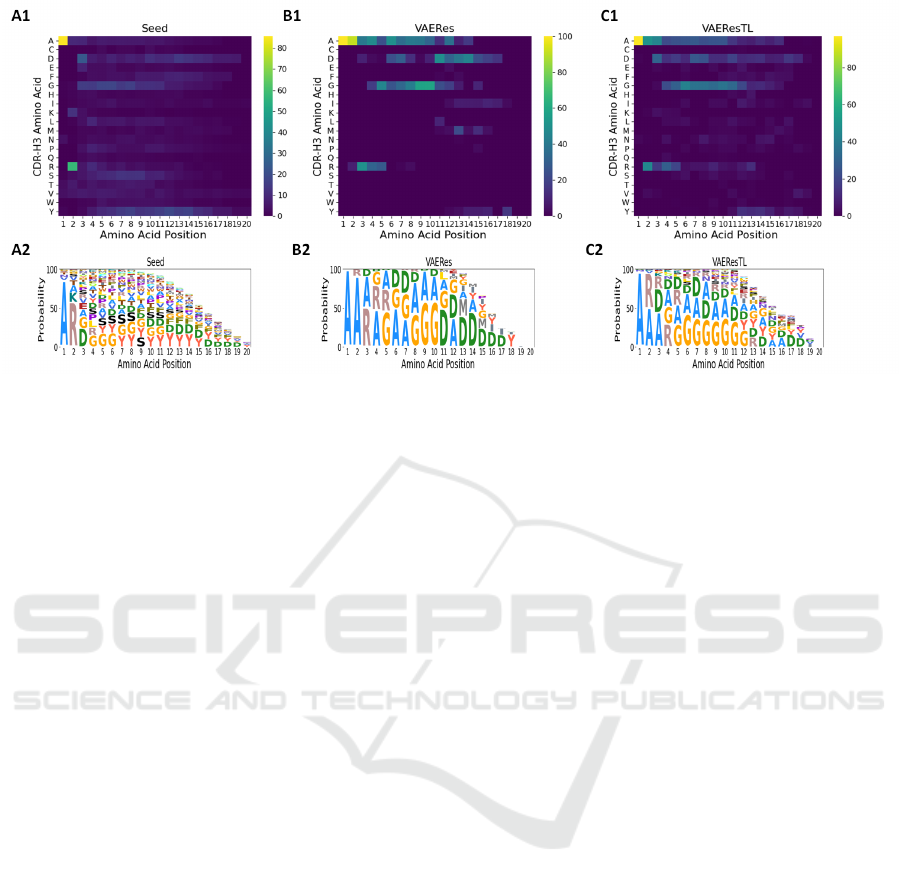
Figure 3: Visualization of machine learning (ML). Position proposed map. Heatmap visualization showing: (A1) the count
of observed seed sequences for SARS-CoV-2. (B1) the count of VAERes-proposed sequences for SARS-CoV-2 from (left)
and to (right) each amino acid at each sequence position. (C1) the count of VAEResTL-proposed sequences for SARS-
CoV-2 from (left) and to (right) each amino acid at each sequence position. (seed and VAERes-generated and VAEResTL-
generated sequences have the length of 20). Sequence logo visualizations. (A2) Sequence logos for Seeds SARS-CoV-2, (B2)
VAERes-generated sequences for SARS-CoV-2 (C2) VAEResTL-generated sequences for SARS-CoV-2 are based on residue
frequency. Sequence logos are computed using Skylign.
in-silico screening to select the best in class lead can-
didates. Although, the predicted sequences are not in
clinical stage yet, we characterized the VAEResTL-
generated SARS-CoV-2 CDR-H3 sequences com-
pared to seed CDR-H3 sequences on a number of
these in-silico methods. In order to screen the CDR-
H3 sequences’ viscosity and clearance we measured
net-charge and hydrophobicity (H) by calculating ev-
ery amino acid sequence of the CDR-H3 sequences.
For all the sequences in the library the net-charge is
calculated at a given pH=7.0 and the hydrophobicity
scale used is “eisenberg” (M
¨
uller et al., 2017). The
optimal net-charge for drug clearance is between 0-
6.2 with H of < 0. Therefore, we filtered out the se-
quences with a net-charge of < 0 (Figure 4A, marked
with red box) and a H of < 0 (Figure 4B, marked
with red box). We also calculated the stability of
CDR-H3 sequences based on proteins and their dipep-
tide composition and filtered out sequences with sta-
bility values of > 40 and < 20 (Figure 4C, marked
with red boxes). We then ran VAEResTL-generated
CDR-H3 amino acid sequences through CamSol to
estimate their solubility. We filtered out sequences
with the CamSol score of < 0.2 (Figure 4D, marked
with red box) according to Sormanni et al. (Sormanni
and Vendruscolo, 2019) guidelines. The low immuno-
genicity of antibodies/nanobodies is an essential bio-
physical property for their therapeutics developabil-
ity. We predicted the peptide binding affinity of all
padded CDR-H3 sequences to MHC Class II by uti-
lizing NetMHCIIpan (Jensen et al., 2018) to reduce
their immunogenicity. We then used peptide’s %Rank
of predicted affinity that we calculated when compar-
ing CDR-H3 sequences with a set of 200,000 random
natural peptides. We predicted affinity for a set of 26
HLA alleles which covers over 98% of the global pop-
ulation. We filtered out the sequences with a %Rank
of < 2.5 (figure 4E, marked with red box) (SARS-
CoV-2 minimum %Rank) and %Rank of < 70 (Fig-
ure 4F, marked with red box) (SARS-CoV-2 average
%Rank). After employing in-silico screening anti-
body/nanobody CDR-H3 variants with desired vis-
cosity, clearance, solubility, stability, and immuno-
genicity remained as potential lead candidates. All
remaining predicted CDR-H3 variants against SARS-
CoV-2, marked outside the red boxes, confined values
equal or superior to the parameters of the SARS-CoV-
2 seed sequences. Through the in-silico screening,
we identified CDR-H3 sequence variants with opti-
mized multi-parameters that can be further evaluated
in a wet-lab setting. However, in our future work
additional filters, including specificity and humaniza-
tion, could be implemented to find the most devel-
opable therapeutic candidates. In addition, mapping
the predicted CDR-H3 sequences in-silico on pro-
tein/epitope targets can be a valuable validation for
our future work.
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
112
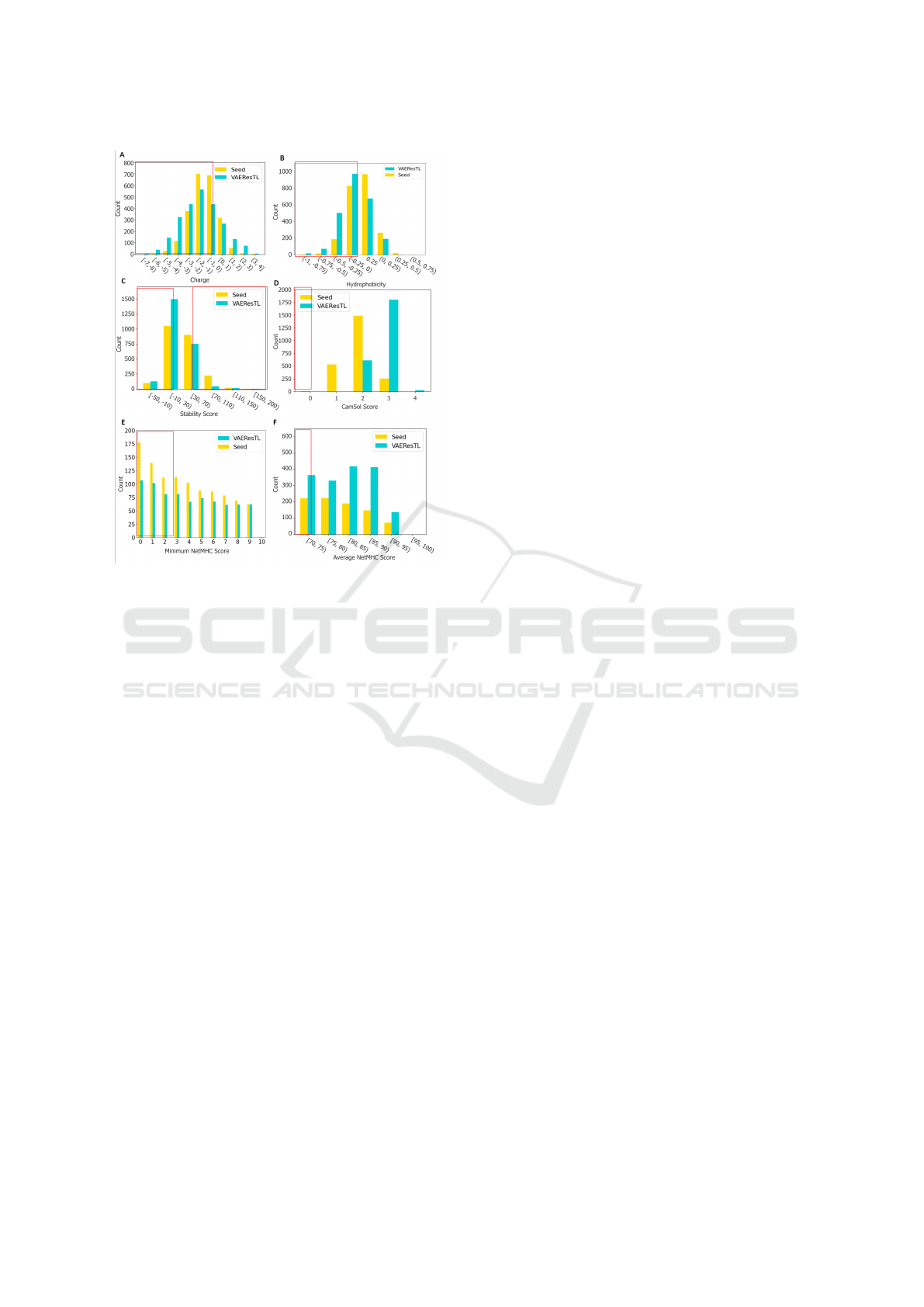
Figure 4: In-Silico Screening of predicted CDR-H3 se-
quences. Histograms present the parameter distributions of
generated and seed SARS-CoV-2 sequences for different fil-
tering steps. Red boxes show filtering cut-off for the ther-
apeutic index in clinical setting. (A) CDR-H3 net-charge.
(B) CDR-H3 H. (C) CDR-H3 stability score. (D) CamSol
solubility score. (E) the minimum NetMHCIIpan %Rank
across all possible 15-mers for a given sequence and across
all 26 HLA alleles. (F) the average NetMHCIIpan %Rank
across all possible 15-mers for a given sequences and across
all 26 HLA alleles.
5 CONCLUSIONS
The results of our study exhibit successful application
of Resnet adopted VAE for generating novel CDR-H3
sequences. We further illustrate how transfer learning
techniques can maximize the power of our VAERes
model for antibody/nanobody discovery when dealing
with the lack of training data for novel targets such as
SARS-CoV-2. Our model was trained on hundreds of
thousands of known and diverse CDR-H3 sequences
from well-studied targets and created a readily usable
tool with extensive generalization capabilities to dis-
cover new antibody/nanobodybased therapeutics. To
select antibodies/nanobodies with improved charac-
teristics, we identified best lead CDR-H3 sequences
with binding ability to SARSCoV- 2 through in-silico
screening. The outcome of this proof-of-concept
study can drive future work for validation of lead can-
didates through wet-lab experiments and the expan-
sion of our model for discovery of other CDR frac-
tions to develop therapeutics against different vari-
ants of SARS-CoV-2 including Delta and Omicron,
as well as other targets. In our future work we will
also employ our VAEResTL to design bispecific and
trispecific antibodies to develop next generation can-
cer therapeutics.
ACKNOWLEDGEMENTS
This wok was supported by MarWell Bio Inc. No ex-
ternal grants or funding contributed to the completion
of this work. All present and future rights to any in-
tellectual property arising from this work will be the
sole property of MarWell Bio Inc.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
Adams, R. M., Mora, T., Walczak, A. M., and Kinney, J. B.
(2016). Measuring the sequence-affinity landscape
of antibodies with massively parallel titration curves.
Elife, 5:e23156.
Adolf-Bryfogle, J., Kalyuzhniy, O., Kubitz, M., Weitzner,
B. D., Hu, X., Adachi, Y., Schief, W. R., and Dun-
brack Jr, R. L. (2018). Rosettaantibodydesign (rabd):
A general framework for computational antibody de-
sign. PLoS computational biology, 14(4):e1006112.
Das, P., Wadhawan, K., Chang, O., Sercu, T., Santos, C. D.,
Riemer, M., Chenthamarakshan, V., Padhi, I., and
Mojsilovic, A. (2018). Pepcvae: Semi-supervised
targeted design of antimicrobial peptide sequences.
arXiv preprint arXiv:1810.07743.
Friedensohn, S., Neumeier, D., Khan, T. A., Csepregi, L.,
Parola, C., de Vries, A. R. G., Erlach, L., Mason,
D. M., and Reddy, S. T. (2020). Convergent selection
in antibody repertoires is revealed by deep learning.
bioRxiv.
Goceri, E. (2019). Analysis of deep networks with resid-
ual blocks and different activation functions: classifi-
cation of skin diseases. In 2019 Ninth international
conference on image processing theory, tools and ap-
plications (IPTA), pages 1–6. IEEE.
Goldstein, L. D., Chen, Y.-J. J., Wu, J., Chaudhuri, S.,
Hsiao, Y.-C., Schneider, K., Hoi, K. H., Lin, Z., Guer-
rero, S., Jaiswal, B. S., et al. (2019). Massively
parallel single-cell b-cell receptor sequencing enables
rapid discovery of diverse antigen-reactive antibodies.
Communications biology, 2(1):1–10.
VAEResTL: A Novel Generative Model for Designing Complementarity Determining Region of Antibody for SARS-CoV-2
113

Guo, X., Tadepalli, S., Zhao, L., and Shehu, A. (2020).
Generating tertiary protein structures via an inter-
pretative variational autoencoder. arXiv preprint
arXiv:2004.07119.
Gupta, A., M
¨
uller, A. T., Huisman, B. J., Fuchs, J. A.,
Schneider, P., and Schneider, G. (2018). Generative
recurrent networks for de novo drug design. Molecu-
lar informatics, 37(1-2):1700111.
He, K., Zhang, X., Ren, S., and Sun, J. (2016). Deep resid-
ual learning for image recognition. In Proceedings of
the IEEE conference on computer vision and pattern
recognition, pages 770–778.
Heinzinger, M., Elnaggar, A., Wang, Y., Dallago, C.,
Nechaev, D., Matthes, F., and Rost, B. (2019). Mod-
eling aspects of the language of life through transfer-
learning protein sequences. BMC bioinformatics,
20(1):1–17.
Jensen, K. K., Andreatta, M., Marcatili, P., Buus, S., Green-
baum, J. A., Yan, Z., Sette, A., Peters, B., and Nielsen,
M. (2018). Improved methods for predicting peptide
binding affinity to mhc class ii molecules. Immunol-
ogy, 154(3):394–406.
Lin, J. (1991). Divergence measures based on the shannon
entropy. IEEE Transactions on Information theory,
37(1):145–151.
Liu, G., Zeng, H., Mueller, J., Carter, B., Wang, Z., Schilz,
J., Horny, G., Birnbaum, M. E., Ewert, S., and Gif-
ford, D. K. (2020). Antibody complementarity de-
termining region design using high-capacity machine
learning. Bioinformatics, 36(7):2126–2133.
Lu, S., Hong, Q., Wang, B., and Wang, H. (2020). Effi-
cient resnet model to predict protein-protein interac-
tions with gpu computing. IEEE Access, 8:127834–
127844.
Mason, D. M., Friedensohn, S., Weber, C. R., Jordi, C.,
Wagner, B., Meng, S., Gainza, P., Correia, B. E., and
Reddy, S. T. (2019). Deep learning enables thera-
peutic antibody optimization in mammalian cells by
deciphering high-dimensional protein sequence space.
BioRxiv, page 617860.
M
¨
uller, A. T., Gabernet, G., Hiss, J. A., and Schneider, G.
(2017). modlamp: Python for antimicrobial peptides.
Bioinformatics, 33(17):2753–2755.
Murphy, K., Travers, P., Walport, M., and Janeway, C.
(2008). Janeway’s Immunobiology - 7th (Seventh) edi-
tion. Garland Science, New York.
Needleman, S. B. and Wunsch, C. D. (1970). A gen-
eral method applicable to the search for similarities
in the amino acid sequence of two proteins. Journal
of molecular biology, 48(3):443–453.
Norman, R. A., Ambrosetti, F., Bonvin, A. M., Colwell,
L. J., Kelm, S., Kumar, S., and Krawczyk, K. (2020).
Computational approaches to therapeutic antibody de-
sign: established methods and emerging trends. Brief-
ings in bioinformatics, 21(5):1549–1567.
Ong, E., Wong, M. U., Huffman, A., and He, Y. (2020).
Covid-19 coronavirus vaccine design using reverse
vaccinology and machine learning. Frontiers in im-
munology, 11:1581.
Papineni, K., Roukos, S., Ward, T., and Zhu, W.-J. (2002).
Bleu: a method for automatic evaluation of machine
translation. In Proceedings of the 40th annual meet-
ing of the Association for Computational Linguistics,
pages 311–318.
Rabiner, L. R. (1989). A tutorial on hidden markov models
and selected applications in speech recognition. Pro-
ceedings of the IEEE, 77(2):257–286.
Raybould, M. I., Kovaltsuk, A., Marks, C., and Deane,
C. M. (2021). Cov-abdab: the coronavirus antibody
database. Bioinformatics, 37(5):734–735.
Raybould, M. I., Marks, C., Krawczyk, K., Taddese, B.,
Nowak, J., Lewis, A. P., Bujotzek, A., Shi, J., and
Deane, C. M. (2019). Five computational developa-
bility guidelines for therapeutic antibody profiling.
Proceedings of the National Academy of Sciences,
116(10):4025–4030.
Ripoll, D. R., Chaudhury, S., and Wallqvist, A. (2021).
Using the antibody-antigen binding interface to train
image-based deep neural networks for antibody-
epitope classification. PLoS computational biology,
17(3):e1008864.
Sharma, V. K., Patapoff, T. W., Kabakoff, B., Pai, S., Hi-
lario, E., Zhang, B., Li, C., Borisov, O., Kelley, R. F.,
Chorny, I., et al. (2014). In silico selection of ther-
apeutic antibodies for development: viscosity, clear-
ance, and chemical stability. Proceedings of the Na-
tional Academy of Sciences, 111(52):18601–18606.
Sormanni, P., Aprile, F. A., and Vendruscolo, M. (2015).
The camsol method of rational design of protein mu-
tants with enhanced solubility. Journal of molecular
biology, 427(2):478–490.
Sormanni, P. and Vendruscolo, M. (2019). Protein solubil-
ity predictions using the camsol method in the study
of protein homeostasis. Cold Spring Harbor perspec-
tives in biology, 11(12):a033845.
Tan, C., Sun, F., Kong, T., Zhang, W., Yang, C., and Liu,
C. (2018). A survey on deep transfer learning. In
International conference on artificial neural networks,
pages 270–279. Springer.
Tsuchiya, Y. and Mizuguchi, K. (2016). The diversity of h
3 loops determines the antigen-binding tendencies of
antibody cdr loops. Protein Science, 25(4):815–825.
Valeri, J. A., Collins, K. M., Ramesh, P., Alcantar, M. A.,
Lepe, B. A., Lu, T. K., and Camacho, D. M. (2020).
Sequence-to-function deep learning frameworks for
engineered riboregulators. Nature communications,
11(1):1–14.
Wang, Y., Yadav, P., Magar, R., et al. (2020). Bio-informed
protein sequence generation for multi-class virus mu-
tation prediction. bioRxiv.
Xu, J., Mcpartlon, M., and Li, J. (2020). Improved protein
structure prediction by deep learning irrespective of
co-evolution information. bioRxiv.
Yoo, D. K., Lee, S. R., Jung, Y., Han, H., Lee, H. K., Han, J.,
Kim, S., Chae, J., Ryu, T., and Chung, J. (2020). Ma-
chine learning-guided prediction of antigen-reactive
in silico clonotypes based on changes in clonal abun-
dance through bio-panning. Biomolecules, 10(3):421.
Zohar, T. and Alter, G. (2020). Dissecting antibody-
mediated protection against sars-cov-2. Nature Re-
views Immunology, 20(7):392–394.
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
114
