
In Silico De Novo Synthesis, Screening, and ADME/T Profiling of
DNA-pA104R Inhibitors as Potential African Swine Fever
Therapeutics
Kim Rafaelle E. Reyes
a
, Timothy Jen R. Roxas
b
, Marineil C. Gomez
c
and Lemmuel L. Tayo
School of Chemical, Biological, and Materials Engineering and Sciences, Mapúa University,
658 Muralla Street, Intramuros, Manila, Philippines
Keywords: African Swine Fever virus, ADME/T, In Silico, De Novo, Drug-likeness, pA104R, Molecular Docking,
Ligand-based Virtual Screening.
Abstract: African Swine Fever Virus (ASFV) is a dsDNA virus causative of the African Swine Fever (ASF) in wild and
domestic hogs. ASF is characterized by hemorrhagic fever, high mortality, and transmissibility. The binding
of the DNA to the pA104R protein mediates viral replication and genome packaging. In the present study, we
generated nine (9) reference compounds that exhibited high docking affinities through de novo computer-
aided drug design (CADD). These compounds were then used as query molecules to find commercially
available drug-like compounds using ligand-based virtual screening (VS). We were able to retrieve 900 hit
compounds that exhibited the same pharmacophoric activities. Then, these hit compounds were subjected to
drug-likeness filtration to identify the most likely to be developed as commercial drugs based on established
parameters. We identified sixty-two (62) drug-like molecules. Molecular docking was then performed to
determine the top five compounds with the highest binding affinities against the target protein. ADME/T
profiling was done on these compounds to assess their pharmacokinetic properties. Compound 8.45 performed
best based on our devised scoring system. This paper shall serve as a good reference in the discovery and
development of anti-ASFV therapeutics.
1 INTRODUCTION
The African Swine Fever Virus (ASFV) is a highly
transmissible virus causative of the African Swine
Fever (ASF) in wild and domestic hogs. Apart from
its swift spread, ASF is characterized by high
mortality rates, to which death is usually observed a
week after the onset of the disease. The identification
of the viral infection is of little difficulty due to the
readily observable symptoms in infected pigs that
include (1) high fever, (2) reduced locomotor
movements, (3) lack of appetite, (4) huddling, (5)
conjunctivitis, (6) diarrhea and vomiting, (7)
somnolence, (8) dyspnea, (9) seizures, and (10) skin
hemorrhages (Blome et al., 2020). This virus's
a
https://orcid.org/0000-0002-2662-209X
b
https://orcid.org/0000-0002-6009-5334
c
https://orcid.org/0000-0001-6238-5709
d
https://orcid.org/0000-0002-0869-2131
transmission and promulgation rely on vector species
such as ticks that primarily target boars found in the
wilderness. ASFV has evolved from a very mild
strain into a highly transmissible virus that threatens
today's swine population (Chen et al., 2021).
Although tremendously virulent to hogs, there is no
risk of this virus being transmitted to humans and
cause the same threats that it poses for pigs. The virus
indirectly impacts society through the economy since
the meat of the domesticated pigs is often a central
ingredient in making food from all ranges of cuisine.
It is therefore of great importance to develop
therapeutics that could eliminate this virus. Up to this
date, there is no commercially available vaccine or
drug to combat ASF in infected animals. The
scientific community has only relied on control
Reyes, K., Roxas, T., Gomez, M. and Tayo, L.
In Silico De Novo Synthesis, Screening, and ADME/T Profiling of DNA-pA104R Inhibitors as Potential African Swine Fever Therapeutics.
DOI: 10.5220/0010774500003123
In Proceedings of the 15th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2022) - Volume 3: BIOINFORMATICS, pages 15-26
ISBN: 978-989-758-552-4; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
15

strategies to confine the virus and prevent its further
spread.
Regarding its structure, ASFV is classified as a
nucleocytoplasmic large DNA virus (NCLDV),
having a genome size of 180,916 base pairs (Alonso
et al., 2018) and an overall virion diameter of 175-215
nm with a 70-100 nm diameter nucleoprotein core
(Blome et al., 2020). The core is surrounded by: (1)
an internal lipid layer, (2) an icosahedral capsid that
is composed of 1892-2172 capsomeres, and (3) the
dispensable lipid envelope (Alonso et al., 2018). This
virus is highly stable in environmental settings and
thrives in most and protein-rich areas. Furthermore, it
can survive in raw meat products at variable
durations. The structure and architecture of the virus
have not been fully elucidated. However, the
pA104R protein of the ASFV has successfully been
studied (Urbano & Ferreira, 2020), and its
crystallographic structure is available (Liu et al.,
2020). This macromolecule is a DNA-binding protein
essential for viral replication and transcription
(Frouco et al., 2017b). ASFV enters the host cell
through endocytosis and micropinocytosis. Then, the
virus uses the pA104R to interact with the host cell's
DNA, leading to the mass manufacture of the viral
parts via gene editing and the completion of the
replicative cycle of the ASFV (Galindo & Alonso,
2017). The structure of the pA104R is characterized
as histone-like since it shares conserved sequence
homology with the other histone-like proteins derived
from bacteria (Frouco et al., 2017a).
In a study by Liu et al., the researchers
successfully elucidated the structure and determined
the binding properties of the pA104R to the host
DNA (i.e., pA104R-DNA complex). Their findings
illustrated the unique binding pattern of pA104R as it
uses its β-ribbon arm and inserts it in the major
groove of the DNA. Furthermore, the researchers
evaluated the ability of the stilbene derivatives, SD1
and SD4, to inhibit viral replication by disrupting the
pA104R-DNA binding in swine macrophages (Liu et
al., 2020). Zhu et al. utilized protein-protein
interaction (PPI) networks to determine the ASFV-
interacting proteins and assessed some commercially
available drugs, such as Polaprezinc and
Geldanamycin, that could potentially bind to some
viral proteins to inhibit the action of the ASFV (Zhu
et al., 2020). A similar study by Mottola et al. helped
unmask the antiviral activity of fluoroquinolones
against the virus (Zhu et al., 2019). Recently, there
have been explorations on the potential of
antimicrobial peptides (AMPs) and their effect on
porcine viruses, including their mechanism of action
(Pen et al., 2020); however, the AMPs used were
already existing ones, and therefore, further
exploration on more efficient peptides must be done.
Although the research on ASF has reached great
strides in the past years, there is still no potential
candidate to eliminate or inhibit the effects of the
virus in question. It is therefore imperative to devise
new strategies that could identify compounds that
could neutralize ASFV.
Drug discovery and development is defined as the
process of identifying chemical entities that have
potential therapeutic effects (Mohs & Greig, 2017).
Over the years, this process has undergone radical
changes with the further integration of biology,
chemistry, physics, mathematics, and computer
science (Umashankar & Gurunathan, 2015). The
pipeline (i.e., drug discovery and development)
involves a multistage process that should be strictly
followed before a novel chemical entity is
commercially available for public consumption
(Mohs & Greig, 2017). Until the late 1980s, drug
discovery was solely based on blind screening and
serendipity (Kiriiri et al., 2020). This was changed
upon introducing high-throughput screening (HTS)
and combinatorial chemistry, allowing scientists to
discover and synthesize many compounds
(Umashankar & Gurunathan, 2015). However, the
methods above were very costly and could be
described as "brute force" approach as finding lead
candidates is dependent on the initial library of
compounds (Polanski, 2020). A further refinement of
the pipeline has emerged with the introduction of the
in silico approach. Such a strategy uses computational
methods to predict the binding properties of the
compound of interest to the biological target. The
Boston Consulting Group estimated that integrating
in silico practices in the drug discovery pipeline could
save 14% of the total cost (Agarwal et al., 2017).
Herein, we identified potential drug-like
compounds that can be utilized to treat African Swine
Fever (ASF) mainly through the inhibition of the
pA104R-DNA binding. To accomplish this, de novo
methods and a ligand-based virtual screening
approach were employed. The binding affinities of
the generated and retrieved compounds were
determined through molecular docking studies.
Finally, the pharmacokinetic properties of the
identified drug-like compounds were ADME/T
studies. This study shall only entail the identification
and pharmacokinetic characterization of the possible
hit compounds.
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
16
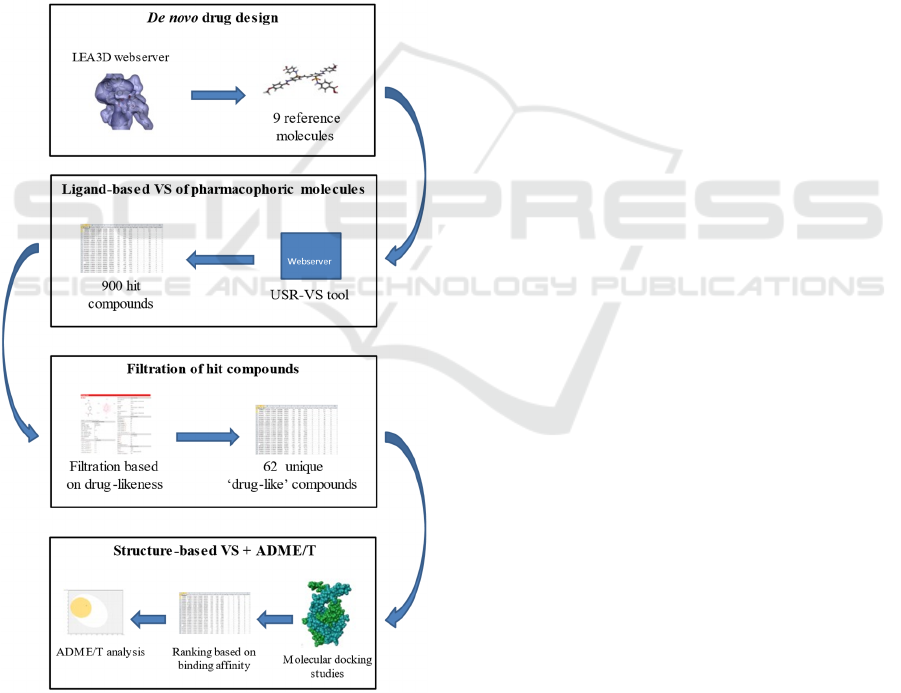
2 METHODOLOGY
2.1 Protein Preparation
The crystallographic structure of ASFV pA104R in
complex with dsDNA was obtained from Protein
Data Bank (accession number: 6LMH). Protein
visualization and refinement were conducted in the
Biovia discovery studio visualizer (DASSAULT
SYSTEMES, 2020). pA104R contains two chains: A
and B (i.e., AHR and BDR, respectively). In this
study, only the AHR was considered, and so the chain
B was removed. The heteroatoms (i.e., dsDNA and
water molecules) were deleted and polar hydrogen
was added for the subsequent analyses. The pA104R-
DNA complex active site residues were shown in
Table 1.
Figure 1: In silico experimental design.
2.2 In Silico De Novo Synthesis of
Reference Molecules
To our knowledge, there has been no definitive report
of possible drug-like DNA-binding inhibitors of the
African swine fever virus. As such, we opted to
perform a de novo drug design approach to generate
potential ligands. This strategy uses computational
algorithms to build molecules that exhibit specified
properties. e-LEA3D (https://chemoinfo.ipmc.cnrs
.fr/LEA3D/index.html) is an online tool that enables
users to perform computer-aided de novo drug design
efficiently. This web server creates diverse
molecules through a genetic algorithm that evolves
fragments based on mutation and crossover operators
(Douguet, 2010).
The prepared protein structure was loaded to the
server. Then, the desired molecular properties of the
output molecules were selected using default
parameters. e-LEA3D uses the PLANTS docking
program to assign scores in the generated molecules.
The parameters used in this function were as follows:
binding site radius = 40, binding site residue =
LYS89, weight in final score = 1. The server returned
10 'reference' compounds that have a high binding
affinity towards pA104R. The experimental design
employed in this study was shown in Figure 1.
2.3 Ligand-Based Virtual Screening
(LBVS)
The de novo approach has one major flaw: molecules
generated through this strategy are hard to synthesize
(Mouchlis et al., 2021). To make up for this
drawback, we adapted ligand-based virtual screening
that could be used to search for commercially
available active compounds from several enormous
libraries of molecules. This approach is usually done
when there is no prior knowledge of the 3D structure
of the target protein (Hamza et al., 2012).
Nonetheless, ligand-based VS could also be
employed when searching for new ligands with
similar chemical and biological activities.
In this study, the USR-VS web server
(http://usr.marseille.inserm.fr/) was used. This tool
implements Ultrafast Shape Recognition (USR) and
Ultrafast Shape Recognition with CREDO Atom
Types (USRCAT) algorithms to screen a library for
similar compounds relative to the pharmacophoric
properties of the query molecule (Schreyer &
Blundell, 2012). Currently, the USR-VS screening
library is comprised of 23 million molecules with
over 94 million low-energy conformers. To conduct
the virtual screening, the structure data files (SDF) of
the nine (9) reference molecules were retrieved from
the e-LEA3D webserver.
In Silico De Novo Synthesis, Screening, and ADME/T Profiling of DNA-pA104R Inhibitors as Potential African Swine Fever Therapeutics
17

The screening process was straightforward. The
SDF files were uploaded into the webserver. Then,
the desired scoring algorithm (i.e. USR and
USRCAT) was selected. We used USRCAT since it
has been reported that this algorithm outperformed
USR in retrospective screening (Schreyer & Blundell,
2012). Clicking 'submit' initiates the virtual screening
that will only take milliseconds. Each query (i.e.
reference) molecule generated 100 hits. Once the
screening was finished, the results were displayed in
a separate internet tab and the SDF for each hit
compound was downloaded.
2.4 Drug-likeness Filtration
Drug-likeness filters are important aspects of drug
discovery. These parameters determined the
likelihood of a compound to exhibit therapeutic
effects while being biologically safe based on its
molecular structure. Moreover, applying filters to a
large library of compounds could eliminate the non-
drug-like molecules, thus saving operation time and
cost (Shen et al., 2012). In this study, a total of 900
hit compounds were identified through ligand-based
virtual screening. Five drug-like filters were
employed for the analysis (Table S1). The filtration
process was conducted using the SwissADME
website (http://www.swissadme.ch/index.php). This
tool is a versatile free-of-charge webserver for
determining the physicochemical properties,
lipophilicity, water-solubility, pharmacokinetics,
drug-likeness, and medicinal chemistry of small
molecules (Daina et al., 2017).
SwissADME only accepts chemical structures in
SMILES format (i.e. .smi extension). Thus, the hit
compounds (i.e., in SDF format) must be converted
into the acceptable file format before executing the
filtration process. To do this, the OpenBabel widget
of the PyRx version 0.8 (The Scripps Research
Institute, 2008) was used. After the said conversion,
the SMILES were uploaded to the webserver. All
compounds that passed the five filtration parameters
without any violations were selected for the
subsequent analyses. Sixty-two (62) unique drug-like
compounds were identified.
2.5 Molecular Docking Studies
The 62 drug-like compounds were subjected to
molecular docking studies to determine their affinity
for binding against pA104R. DockThor
(https://dockthor.lncc.br/v2/) is a user-friendly
webserver for receptor-ligand docking developed by
the GMMSB group (Santos et al., 2020). This tool
performs molecular docking through flexible-ligand
and rigid-receptor strategies based on the MMFF94S
force field (Guedes, Costa, et al., 2021). The docking
procedure is a three-step process. First, the prepared
protein (i.e. protonated) in PDB format was uploaded
to the server. Since no cofactors were considered in
this study, the 'do not use cofactor' function was
selected. Then, the 62 drug-like compounds in SDF
format were docking program requires the user to
upload the protonated version of the ligands. For
convenience, DockThor is embedded with an 'add H'
function. The submitted protein and ligand structure
were processed after clicking 'send'. A checkmark
appeared which indicated that the input molecules
were valid and recognized by the force field. The final
step involves setting up the docking configuration.
The user could choose from blind docking or user-
defined docking. Since the binding site was already
determined (Table 1), we performed user-defined
docking. DockThor utilizes a genetic algorithm to
determine the optimal poses for flexible ligand
docking (De Magalhães et al., 2014). Furthermore,
the platform allows the user to customize the
algorithm parameters, but the 'standard algorithm'
was selected for this study. Table S2 shows the
different parameters used in the docking experiment.
The webserver ranked the 62 drug-like compounds
based on their binding affinities. The chemical
structure (i.e., in .mol2 format) of the best docking
pose for each input molecule was obtained. The 3D
and 2D protein-ligand interactions were visualized
using Biovia discovery studio.
2.6 ADME/T Studies
Compounds must undergo ADME/T studies to
determine their pharmacokinetic properties and
safety level. The 62 drug-like compounds and the 9
reference molecules were then subjected to ADME/T
studies. To do so, we used pkCSM
(biosig.unimelb.edu.au/pkcsm/prediction), a web-
based tool commonly used to calculate the
pharmacokinetic properties of small-molecule drugs,
such as the compounds involved in this study. This
application allows for the fast development of
predictive models of central ADMET properties via
graph-based signatures (Pires et al., 2015). Since the
pkCSM webserver only accepts entries in the
SMILES format, we first converted the available files
to the .smi format via the OpenBabel widget of the
PyRx software, similar to the earlier method.
Subsequently, the compounds were uploaded to the
web-based server for the prediction of their
pharmacokinetic properties
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
18

3 RESULTS AND DISCUSSION
3.1 The pA104R: A Therapeutic Target
for ASF
DNA packaging is a vital process in the life cycle of
double-stranded DNA (dsDNA) viruses. Packaging
proteins bind with the DNA and initiates
conformational changes that cause it to bend and be
organized into densely packed chromatin structures
(Urbano & Ferreira, 2020). Failure of these proteins
to promote condensation and packaging will
inevitably cause DNA damage, ultimately leading to
apoptosis (J. Y. J. Wang, 2001). Therefore, targeting
the proteins involved in the said process is an
attractive approach to design therapeutics against
viruses. DNA-packaging proteins have been reported
for a wide range of organisms. For instance, the
packaging proteins in bacteria are the histone-like
proteins belonging to the HU/IHF superfamily
(Swinger & Rice, 2004). In relevance to the ASFV,
p10 and pA104R are the major DNA-packaging
proteins in mature ASFV (Andrés et al., 2002).
However, a more recent study using small interfering
RNA (siRNA) has shown that pA104R has a
profound role in DNA replication, transcription, and
packaging of ASFV (Frouco et al., 2017a).
Figure 2: The structure of (A) DNA-pA104R complex
(PDB accession code:6LMJ) and (B) apo-pA104R (PDB
accession: 6LMH).
pA104R is a homodimer that significantly
resembles other bacterial HU/IHF homologs (Liu et
al., 2020). The crystallographic structures of the apo-
pA104R and DNA-bound pA104R were shown in
Figure 2. The protein folds into two domains, namely
the "body" AHR(i.e., alpha-helical region) and the
"arms" BDR (i.e., β-strand DNA binding region). As
shown in Fig 2B, the DNA interacts predominantly
with pA104R via the BDR arm. The surface of this
region is saturated with positively charged, making it
an attractive binding site for the negatively charged
DNA molecule (Liu et al., 2020). Thus, the
subsequent analyses were simplified by focusing on
the BDR. To design an effective inhibitor, the key
amino acid residues within the binding site must be
identified. Thus, the active site amino acid residues in
the DNA-pA104R complex were determined using
the Biovia Discovery Studio (Table 1). As
hypothesized, the key amino acid residues in the BDR
arm are mostly positively charged at physiological
pH. HIS78, LYS89, and LYS91 are all positively
charged. Meanwhile, PRO80 is an aliphatic amino
acid making it nonpolar and hydrophobic. This
residue interacts with the DNA strand, but the nature
of its interaction is not electrostatic since it is
nonpolar at physiological pH. Further studies are
encouraged to uncover the linkages between this
residue and the target DNA strand.
Table 1: Interacting amino acid of DNA-pA104R complex.
Only the AAs in the BDR were considered.
p
A104R domain Active residues
AHR
LYS 63
LYS98
ARG100
LEU102
LYS103
BDR
HIS78
PRO80
LYS89
LYS91
3.2 De Novo CADD of ASFV DNA
Binding Inhibitors
There is only a handful of literature dedicated to
searching for possible DNA-binding inhibitors in
AFSV. Liu et al. reported that SD1 and SD4 (i.e.,
stilbene derivatives) had inhibitory effects on the
DNA-pA104R binding. Such results were evident by
their ability to repress the ASFV replication (Liu et
al., 2020). To our knowledge, these are the only
molecules known to have therapeutic potential
against African swine fever. It is of great importance
to increase the number of viable ligands. De novo
computer-aided drug design (CADD) approach in
drug discovery enables the generation of novel
ligands based on defined scoring functions (Douguet,
2010). To that end, e-LEA3D, a de novo drug design
tool, was employed in this study. This program uses
a genetic algorithm that evolves molecular fragments
and optimizes the combination of these fragments
(Douguet et al., 2005). Once a library of optimized
molecules is generated, they are assigned a score
based on docking fitness calculated by the PLANTS
program. As shown in Table 2, e-LEA3D generated
nine molecules. ref1 has the highest score (i.e.,
91.25%), implying that this compound has the best
docking conformation from all the generated
molecules using the program's algorithm. However,
In Silico De Novo Synthesis, Screening, and ADME/T Profiling of DNA-pA104R Inhibitors as Potential African Swine Fever Therapeutics
19

this does not bear weight on the binding affinity of the
ligand to pA104R since its primary purpose is to rank
the solutions.
Table 2: Reference molecules generated using e-LEA3D.
Code
Molecular
Formula
Weight
(g
mol
-1
)
Score
(
%
)
ref1 C
48
H
61
N
10
O
14
S
2
1066.19 91.25
ref2 C
68
H
86
N
10
O
17
S
3
1411.66 87.07
ref3 C
43
H
57
N
11
O
12
S
2
984.11 84.89
ref4 C
67
H
84
N
10
O
17
S
3
1397.64 84.31
ref5 C
67
H
82
N
14
O
17
S
3
1451.65 83.75
ref6 C
55
H
64
N
10
O
12
S
2
1121.29 83.31
ref7 C
62
H
82
N
10
O
17
S
3
1335.57 83.29
ref8 C
62
H
82
N
10
O
17
S
3
1335.57 83.23
ref9 C
49
H
60
N
10
O
16
S
3
1141.25 82.94
3.3 Ligand-Based Virtual Screening
(LBVS) of Molecules Based on
Pharmacophoric Activity
The de novo design strategy of molecules does not
guarantee their ability to be developed into
therapeutic agents. As mentioned, the synthetic
accessibility of the generated compounds is one of the
major challenges in the de novo approach (Mouchlis
et al., 2021). A high binding affinity serves no
purpose if the molecule is hard to synthesize. To
address this problem, a screening process within a
library of commercially available molecules may be
performed. Virtual screening (VS) of prospective
drug compounds has become the norm in the early
stages of drug discovery. It is often regarded as the in
silico counterpart of the tedious and expensive high-
throughput screening (HTS) (Polgar & M. Keseru,
2011).
The screening process is divided into ligand-based
and structure-based approaches. The latter aims to
determine the best ligand that will bind to the receptor
based on surface complementarity (Maia et al., 2020).
The pre-requisite for this type of analysis is the
availability of the 3D protein structure. Meanwhile,
ligand-based VS uses the pharmacophoric properties
of a query molecule to retrieve compounds that
exhibit similar biological and chemical activities
from a database(Singh et al., 2021).
In this study, we applied ligand-based virtual
screening to obtain commercially available molecules
based on the pharmacophores of the reference
compounds. USR-VS is a webserver that uses
Ultrafast Shape Recognition (USR), and Ultrafast
Shape Recognition with CREDO Atom Types
(USRCAT) algorithms for effective pharmacophore
search and retrieval (Li et al., 2016). USR predicts the
molecular shape by analyzing the relative positions of
bonded atoms. As implied by its name, USR enables
the user to search for molecules with a similar three-
dimensional shape at incredible speed. USRCAT is
an extension of USR integrated with the CREDO, a
structural interactomics database (Schreyer &
Blundell, 2013). This algorithm works similarly with
USR, but it uses pharmacophoric constraints for a
more effective similarity search. Therefore, the
USRCAT algorithm was used in this analysis. The
nine (9) de novo designed compounds were used as
query molecules to the USR-VS webserver. The
similarity search covered 23 million molecules and 94
million low-energy conformers from the ZINC
database. Each run returned 100 hit compounds.
Therefore, the nine reference molecules generated
900 hits.
3.4 Drug-likeness Filtration of Hit
Compounds
It is estimated that only 40% of hit compounds can
transition from the pre-clinical to first-in-humans
stage due to their poor physical and chemical
properties (Venkatesh and Lipper, 2000). Drug-
likeness filtration is one of the barriers a compound
must overcome to advance in the late phases of drug
discovery (Hu et al., 2018). This assesses the
probability of a compound to be manufactured as a
therapeutic drug based on some physicochemical
parameters. The method of applying the drug-likeness
filter has been an integral step in the drug discovery
pipeline because any chemical compound may
exhibit an excellent therapeutic effect. Still, not all
could be transformed into viable drug.
To eliminate the hit compounds with undesirable
properties, drug-likeness filtration was performed
using the SwissADME webserver. This web tool has
been used in 2100 in silico analyses (i.e., as per the
number of citations of the published paper of the
developers (Daina et al., 2017) ). SwissADME uses
five filters to assess the drug-like properties.
Violation in any of the filters (i.e., Lipinski (Lipinski,
2004), Ghose (Ghose et al., 1999), Egan (Egan et al.,
2000), Veber (Veber et al., 2002), and Muegge
(Muegge et al., 2001)) disqualifies the compound
from further analysis. By adhering to this selection
criterion, one could ascertain the excellent drug-like
properties of successful compounds.
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
20

Table 3: Drug-likeness filtration of the 900 compounds.
(Note: For simplification, only results from five compounds
were shown).
Compound
no.
Code Formula
No. of filter
violations
(i.e., Lipinski, Ghose,
Veber, Egan, and
Muegge)
1
1.27 C
23
H
30
N
4
O
2
0
10
2.59 C
19
H
23
N
3
O
3
0
54
9.19 C
20
H
31
N
3
O
2
S 0
58
9.43 C
20
H
22
N
2
O
3
0
62
9.92 C
24
H
26
N
2
O
4
0
Table 3 shows the summary of the results. After
screening 900 compounds, only 62 were drug-like.
This translates to a 6.89% success rate from hit
identification to drug-like filtration. Lipinski's rule of
five (Ro5) was primarily developed to assess the
druggability of new molecular entities (Lipinski,
2000). If a molecule fails one of the parameters of
Ro5, then the absorption and permeability properties
are put into question. However, Lipinski et al. stated
that molecules that violate at least one of the said
parameters should not be necessarily removed from
the selection process (Petit et al., 2012). Instead, such
molecules should be given low priority in the drug
discovery process. Nonetheless, satisfying the Ro5
without violation is an indicator of excellent drug-
likeness.
3.5 Molecular Docking Studies
Sixty-two (62) identified drug-like molecules
underwent further screening to determine the
compounds that exhibit high binding interactions
with the target protein pA104R. The analysis was
conducted through molecular docking, a structure-
based virtual screening strategy. Molecular docking
is a computational approach to screen for ligands that
fit the protein's ligand-binding site with high
complementarity (i.e., geometrically and
energetically) (Azam & Abbasi, 2013). Docking
tools use search algorithms to predict a ligand's best
docking pose (Sanchez, 2013). Then, a scoring
function calculates the binding free energy of the
protein-ligand complex (Bissantz et al., 2000).
In this study, DockThor, a web server for highly
flexible ligand-docking, was used. This tool utilizes a
dynamic genetic algorithm as a search method. Such
an algorithm allows the intensive survey of the energy
hypersurface to generate multiple minima solutions
(De Magalhães et al., 2014). DockThor uses the
DockTScore as a scoring function based on the
MMFF94S force field (Guedes, Barreto, et al., 2021).
The scoring function considers the intermolecular
interactions, torsional entropy, lipophilic interaction,
polar solvation, and nonpolar solvation. As shown in
Table 4, the binding affinities achieved range from -
7.790 to -6.158 kcal mol
-1
. Compound 8.45 ranked
first with a binding affinity of -7.790 kcal mol
-1
.
Table 4: Docking results of the 62 drug-like compounds.
(Note: For simplification, only results from five compounds
were shown).
Rank
Compound
Code
Binding
affinity
(kcal mol
-1
)
Total
energy
(kcal mol
-1
)
vdW
Energy
Elec.
energy
1 8.45 -7.790 12.623 -15.214 -11.853
2 2.21 -7.705 36.308 -13.926 -11.001
52 2.59 -6.817 18.362 -4.727 -19.801
51 9.19 -6.841 -1.480 -8.951 -16.563
62 6.45 -6.158 -7.153 -2.613 -19.790
There are two amino acids critical for the binding
of 8.45 with ASFV. GLN76 (i.e., glutamine at
position 76) formed three hydrogen bonds, two of
which are conventional, while the remaining is a pi-
donor hydrogen bond. The first hydrogen bond is
formed by the interaction of the oxygen atom of the
GLN76 to the hydrogen atom of the amino group in
8.45. Then, the hydrogen from the GLN76 interacts
with the carbonyl oxygen of the compound.
Meanwhile, the glutamine's nitrogen atom forms a pi-
donor hydrogen bonding (Figure 3). HIS78 creates a
pi-alkyl interaction with the said molecule. The
amino acid, HIS78, is one of the active site amino acid
residues (Table 1). Such interactions might explain
why compound 8.45 had the highest binding affinity
among all drug-like molecules that underwent
molecular docking. Therefore, 8.45 could be a
Figure 3: Molecular interactions of compound 8.45 to (top)
GLN76 and (bottom) HIS78 of the ASFV.
In Silico De Novo Synthesis, Screening, and ADME/T Profiling of DNA-pA104R Inhibitors as Potential African Swine Fever Therapeutics
21

potential DNA-binding inhibitor of pA104R solely
based on the docking results. However, further tests
must be conducted to determine this compound's
potential as a therapeutic agent.
3.6 ADME/T Profiling
Determination of pharmacokinetic characteristics is
one of the most critical steps to ensure that the drug
being developed is safe to be administered during
animal and clinical trials. The results for the ADME/T
of the unique compounds with the highest binding
affinities are shown in Table 5. For the absorption
parameters, it can be observed that the intestinal
absorption% for the drug-like compounds have a very
high positive value, ranging from 69.985% to
95.646%. Such results match the existing literature
since adhering to Lipinski's Rule of Five entails that
the drug-like compounds are likely to be well-
absorbed in the intestine (Zhao et al., 2001). Further
supporting this idea, the range derived for the human
intestinal absorption (HIA) % was above the optimum
level of 30%, as shown by Wang (2016) (N. N. Wang
et al., 2017). All the values for skin permeability of
both the unique compounds indicate skin
permeability because all the values were lower than -
2.5 (Hassan et al., 2018). The results are favorable
since they signify that the drugs can be applied
through skin contact and promote the elimination of
these drugs to prevent the accumulation of chemicals
in the body (Osborne & Musakhanian, 2018). This
finding provides an alternative route of
administration for the proposed compounds.
Caco-2 permeability is considered the final
absorption parameter. It makes use of the Caco-2 cell,
or the human colon adenocarcinoma, to model the
intestinal absorption of many compounds
(Matsumoto et al., 2018). The Caco-2 permeability
values for the unique group were varying. The unique
and reference groups yielded acceptable results as all
values were above the reported threshold for optimum
Caco-2 permeability value (i.e., -5.15) (N. N. Wang
et al., 2016). This finding reinforces the results given
by the HIA% that the compounds under study have
adequate intestinal absorption.
We observed that the VD
ss
values of the unique
group varied but were negative for all reference
compounds for the distribution parameters. A higher
VD
ss
value entails better distribution into the tissues
than in the plasma (Yates & Arundel, 2008).
Compounds 2.2 and 9.19 had unfavorable VD
ss
values because they were close to the minimum range
(i.e., -0.15) (Firdausy et al., 2020). On the other hand,
the drug compounds 8.45, 2.21, 8.40, 7.21, and 2.59
displayed moderate VD
ss
values because their values
were between the range reported (i.e., -0.15 to 0.45)
(Firdausy et al., 2020).
The blood-brain barrier permeability was varying
for the unique group but all negative for the reference
compounds. Nevertheless, all the unique compounds
were unable to penetrate the blood-brain barrier (i.e.,
< 0.3) (Firdausy et al., 2020). Such a result is a
positive indication since the expected target of the
compounds is not found within the brain. Regarding
CNS permeability, compound 7.21 can effectively
penetrate the central nervous system (i.e., > -2),
whereas the remaining unique compounds could only
poorly penetrate the CNS (i.e., < -3) (Pires et al.,
2015). However, even if the CNS permeability
values were favorable for all the compounds, they
would still not penetrate the CNS due to the blood-
brain barrier (Carpenter et al., 2014).
The metabolism of the compounds being studied
was dictated by their capacity to become either
CYP2D6 or CYP3A4 (i.e., the two main subtypes of
cytochrome P450) inhibitors (Firdausy et al., 2020).
All of the unique compounds were not CYP2D6
inhibitors. Meanwhile, compounds 2.2, 8.40, and
7.21 were known to be CYP3A4 inhibitors. A
negative result from these tests could suggest the
excellent metabolism of the proposed drug-like
compounds in the human body; the presence of
inhibitors poses a threat for the body since decreased
metabolism leads to the accumulation of the
compounds and will thus increase the toxicity of that
potential drug (Niel et al., 1992).
For the excretion parameter, total clearance was
considered. This parameter measures the compound's
ability to be cleared from all tissues (i.e., the
combination of renal and hepatic clearances. The
CLtot values for compounds considered were within
the range -0.278 to 1.449 log ml min
-1
kg
-1
. It was
found that the highest total clearance was achieved by
compound 8.45, which suggests that it has the highest
bioavailability (Firdausy et al., 2020). Meanwhile,
compounds 7.21 and 9.19 had negative values
indicating their poor systemic clearance.
Finally, the toxicity of the proposed drugs was
evaluated. The Ames test is a preliminary evaluation
to determine the mutagenicity of drug candidates
(Mortelmans et al., 2016). Based on the results, only
compound 2.2 was characterized as a mutagen. There
is a high correlation between carcinogenicity and
mutagenicity (ca. 90%). This indicates that
compound 2.2 could induce mutations leading to
cancer (Mortelmans et al., 2016). It is therefore
essential to perform other tests to determine its
genotoxicity.
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
22

Table 5: ADME/T Results of the unique (i.e., drug-like) group. The top 5 compounds based on their binding affinities from
the molecular docking studies were used. Compounds 9.19 and 2.59 were also included for the ADME/T profiling due to
their low hepatoxicity.
Code
Absorption Distribution Metabolism Excretion Toxicity
Intestina
l abs.
Skin
permeability
Caco-2
permeability
VD
ss
BBB
permeabili
ty
CNS
permeabi
lity
CYP 2D6
inhibitor
CYP 3A4
inhibitor
Total
Clearance
Ames
Toxicity
Hepato
toxicity
LD50
(mg kg
-1
)
8.45 89.927 -2.849 0.794 0.361 -0.569 -2.641 No No 1.022 No Yes 695.29
2.21 91.148 -3.022 0.811 0.395 -0.802 -2.653 No No 0.808 No Yes 911.54
2.2 82.316 -2.741 0.158 -0.182 -1.053 -2.42 No Yes 0.109 Yes Yes 1056.52
8.40 92.54 -3.069 0.662 0.221 -0.955 -2.974 No Yes 0.248 No Yes 1081.99
7.21 92.942 -3.246 0.703 0.126 -0.183 -1.953 No Yes -0.317 No Yes 793.17
9.19 91.444 -3.869 0.878 -0.087 -0.375 -2.918 No No -0.278 No No 390.81
2.59 88.437 -2.863 0.987 0.105 -0.836 -2.44 No No 0.438 No No 2583.83
Meanwhile, a hepatotoxicity test was also
performed to determine whether the drug could cause
significant liver injury. This stage of the development
process greatly impedes the translation of a substance
into a commercial drug (Björnsson, 2016). Based on
the results, only compounds 9.19 and 2.59 were not
hepatoxic. This result signifies that these are the only
compounds from the unique group that causes
minimal harm to the human liver.
The final parameter considered is the rat oral acute
toxicity (LD
50
) of the proposed drug candidates. This
parameter determines the amount of the substance
that could kill 50% of the test animal population
(Adamson, 2016). The higher the LD
50
value of the
compound, the less toxic the substance is when taken
orally by the individual. The resulting LD50 of the
unique group compounds ranged from 2.121 to 2.985.
Based on the Hodge and Sterner scale, all compounds
except 9.19 are considered only slightly toxic, with a
toxicity rating of 4 (ca. 500-5000 mg kg
-1
) (Ahmed,
2015). Meanwhile, compound 9.19 is considered
moderately toxic since its LD
50
value falls under a
toxicity rating of 3 (ca. 50-500 mg kg
-1
). The
remaining compounds had relatively low LD
50
that is
indicative of their high toxicity. Therefore, caution
must be exercised when deriving the optimum dosage
of these drug candidates.
To identify the most suitable compounds among
the unique group, we devised a scoring system that
consisted of the molecular docking rank and the
ADME/T score. The top five compounds from the
molecular docking studies were analyzed for
ADME/T profiling. However, we also considered
compounds 9.19 and 2.59 even though they ranked
51
st
and 52
nd
, respectively. These two were the only
non-hepatoxic compounds; therefore, we opted to
include them in the scoring system. For the ADME/T
scoring, each parameter violation was awarded one
point. The compound with the lowest score was
deemed to have the most favorable ADME/T
properties. Based on the results, compound 2.59
performed best on the ADME/T studies (Table 6).
Meanwhile, compound 2.2 had the most number of
parameter violations.
Table 6: Summary of docking and ADME/T performance.
For ADME/T profiling, violation in any of the parameters
is rewarded one point. The final score is the average of the
docking and ADME/T scores.
Code
Docking
Rank/Score
ADME/T Score Final Score
8.45 1 2 1.5
2.21 2 2 2
2.2 3 4 3.5
8.40 4 3 3.5
7.21 5 3 4
9.19 51 2 26.5
2.59 52 1 26.5
The docking score and the ADME/T score were
averaged to calculate the final score. Compound 8.45
had the lowest score (ca. 1.5), signifying its excellent
binding affinity against pA104R and favorable
pharmacokinetic properties. Although compound
2.59 ranked first in the ADME/T studies, it fell short
of its molecular docking ranking resulting in a low
final score (ca. 26.5). Compounds 2.21, 2.2, 8.40, and
7.21 had relatively good final scores. Such results
implied their superior properties similar to compound
8.45. Additional tests must be conducted on these
compounds to determine their capabilities as
therapeutic agents against ASF. Particularly,
bioassays such as the haemadsorption test (HAT) are
useful in exploring the efficiency of the compounds
as ASFV therapeutics (Fischer et al., 2020).
In Silico De Novo Synthesis, Screening, and ADME/T Profiling of DNA-pA104R Inhibitors as Potential African Swine Fever Therapeutics
23

4 CONCLUSION
In this paper, the identification and characterization
of potential drug candidates for the treatment of ASF
were conducted. We were able to characterize the
structure of the pA104R protein with visualization
software. The DNA binding site of the pA104R was
determined. Nine (9) de novo reference compounds
were generated. Of these compounds, 900
commercially available drug-like small molecules
were retrieved through ligand-based virtual screening
using pharmacophoric similarities.
Drug-likeness filtration was done to determine the
compounds with excellent druggability properties.
Sixty-two (62) drug-like compounds were subjected
to molecular docking and ADME/T studies. Of these
filtered drug-like molecules, compound 8.45
achieved exceptional docking rank (ca. 1) and
ADME/T score (ca. 2), earning the lowest final score.
The other drug-like molecules (i.e., 2.21, 2.2, 8.40,
and 7.21) also performed well. Compounds 9.19 and
2.59 had the best ADME/T profile but performed
poorly in the molecular docking studies. Further
experiments must be performed to identify their
potential as anti-ASF therapeutics.
REFERENCES
Adamson, R. H. (2016). The acute lethal dose 50 (LD50) of
caffeine in albino rats. Regulatory Toxicology and
Pharmacology, 80, 274–276. https://doi.org/10.1016/
j.yrtph.2016.07.011
Agarwal, D., Udoji, M., & Trescot, A. (2017). Genetic
Testing for Opioid Pain Management: A Primer. Pain
and Therapy, 6, 1–13. https://doi.org/10.1007/s40122-
017-0069-2
Ahmed, M. (2015). Acute Toxicity (Lethal Dose 50
Calculation) of Herbal Drug Somina in Rats and Mice.
Pharmacology & Pharmacy, 06(03), 185–189.
https://doi.org/10.4236/pp.2015.63019
Alonso, C., Borca, M., Dixon, L., Revilla, Y., Rodriguez,
F., & Escribano, J. M. (2018). ICTV virus taxonomy
profile: Asfarviridae. Journal of General Virology,
99(5), 613–614. https://doi.org/10.1099/jgv.0.001049
Andrés, G., Alejo, A., Salas, J., & Salas, M. L. (2002).
African Swine Fever Virus Polyproteins pp220 and
pp62 Assemble into the Core Shell. Journal of
Virology, 76(24), 12473–12482. https://doi.org/10.112
8/jvi.76.24.12473-12482.2002
Azam, S. S., & Abbasi, S. W. (2013). Molecular docking
studies for the identification of novel melatoninergic
inhibitors for acetylserotonin-O-methyltransferase
using different docking routines. Theoretical Biology
and Medical Modelling, 10(1), 1–16. https://doi.org/
10.1186/1742-4682-10-63
Bissantz, C., Folkers, G., & Rognan, D. (2000). Protein-
based virtual screening of chemical databases. 1.
Evaluation of different docking/scoring combinations.
Journal of Medicinal Chemistry, 43(25), 4759–4767.
https://doi.org/10.1021/jm001044l
Björnsson, E. S. (2016). Hepatotoxicity by drugs: The most
common implicated agents. International Journal of
Molecular Sciences, 17(2). https://doi.org/10.3390/ijms
17020224
Blome, S., Franzke, K., & Beer, M. (2020). African swine
fever – A review of current knowledge. Virus Research,
287(April), 198099. https://doi.org/10.1016/j.virusres.
2020.198099
Carpenter, T. S., Kirshner, D. A., Lau, E. Y., Wong, S. E.,
Nilmeier, J. P., & Lightstone, F. C. (2014). A Method
to Predict Blood-Brain Barrier Permeability of Drug-
Like Compounds Using Molecular Dynamics
Simulations. Biophysical Journal, 107(3), 630–641.
https://doi.org/10.1016/j.bpj.2014.06.024
Chen, S., Zhang, X., Nie, Y., Li, H., Chen, W., Lin, W.,
Chen, F., & Xie, Q. (2021). African Swine Fever Virus
Protein E199L Promotes Cell Autophagy through the
Interaction of PYCR2. Virologica Sinica, 36(2), 196–
206. https://doi.org/10.1007/s12250-021-00375-x
Daina, A., Michielin, O., & Zoete, V. (2017). SwissADME:
A free web tool to evaluate pharmacokinetics, drug-
likeness and medicinal chemistry friendliness of small
molecules. Scientific Reports, 7(January), 1–13.
https://doi.org/10.1038/srep42717
De Magalhães, C. S., Almeida, D. M., Barbosa, H. J. C., &
Dardenne, L. E. (2014). A dynamic niching genetic
algorithm strategy for docking highly flexible ligands.
Information Sciences, 289(1), 206–224.
https://doi.org/10.1016/j.ins.2014.08.002
Douguet, D. (2010). e-LEA3D: A computational-aided
drug design web server. Nucleic Acids Research,
38(SUPPL. 2), 615–621.
https://doi.org/10.1093/nar/gkq322
Douguet, D., Munier-Lehmann, H., Labesse, G., & Pochet,
S. (2005). LEA3D: A computer-aided ligand design for
structure-based drug design. Journal of Medicinal
Chemistry, 48(7), 2457–2468. https://doi.org/10.1021
/jm0492296
Egan, W. J., Merz, K. M., & Baldwin, J. J. (2000).
Prediction of drug absorption using multivariate
statistics. Journal of Medicinal Chemistry, 43(21),
3867–3877. https://doi.org/10.1021/jm000292e
Firdausy, A. F., Muti’ah, R., & Rahmawati, E. K. (2020).
Predicting Pharmacokinetic Profiles of Sunflower’s
(Helianthus annuus L.) Active Compounds using in
Silico Approach. Journal of Islamic Medicine, 4(1), 1–
7. https://doi.org/10.18860/jim.v4i1.8840
Fischer, M., Hühr, J., Blome, S., Conraths, F. J., & Probst,
C. (2020). Stability of African swine fever virus in
carcasses of domestic pigs and wild boar
experimentally infected with the ASFV “Estonia 2014”
isolate. Viruses, 12(10). https://doi.org/10.3390/v12
101118
Frouco, G., Freitas, F. B., Coelho, J., Leitão, A., Martins,
C., & Ferreira, F. (2017a). crossm. 91(12), 1–14.
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
24

Frouco, G., Freitas, F. B., Coelho, J., Leitão, A., Martins,
C., & Ferreira, F. (2017b). DNA-Binding Properties of
African Swine Fever Virus pA104R, a Histone-Like
Protein Involved in Viral Replication and
Transcription. Journal of Virology, 91(12), 1–14.
https://doi.org/10.1128/jvi.02498-16
Galindo, I., & Alonso, C. (2017). African swine fever virus:
A review. Viruses, 9(5). https://doi.org/10.3390/v905
0103
Ghose, A. K., Viswanadhan, V. N., & Wendoloski, J. J.
(1999). A knowledge-based approach in designing
combinatorial or medicinal chemistry libraries for drug
discovery. 1. A qualitative and quantitative
characterization of known drug databases. Journal of
Combinatorial Chemistry, 1(1), 55–68.
https://doi.org/10.1021/cc9800071
Guedes, I. A., Barreto, A. M. S., Marinho, D., Krempser,
E., Kuenemann, M. A., Sperandio, O., Dardenne, L. E.,
& Miteva, M. A. (2021). New machine learning and
physics-based scoring functions for drug discovery.
Scientific Reports, 11(1), 1–19.
https://doi.org/10.1038/s41598-021-82410-1
Guedes, I. A., Costa, L. S. C., dos Santos, K. B., Karl, A. L.
M., Rocha, G. K., Teixeira, I. M., Galheigo, M. M.,
Medeiros, V., Krempser, E., Custódio, F. L., Barbosa,
H. J. C., Nicolás, M. F., & Dardenne, L. E. (2021). Drug
design and repurposing with DockThor-VS web server
focusing on SARS-CoV-2 therapeutic targets and their
non-synonym variants. Scientific Reports, 11(1), 1–20.
https://doi.org/10.1038/s41598-021-84700-0
Hamza, A., Wei, N. N., & Zhan, C. G. (2012). Ligand-based
virtual screening approach using a new scoring
function. Journal of Chemical Information and
Modeling, 52(4), 963–974. https://doi.org/10.1021/ci
200617d
Hassan, M., Shahzadi, S., Seo, S. Y., Alashwal, H., Zaki,
N., & Moustafa, A. A. (2018). Molecular docking and
dynamic simulation of AZD3293 and solanezumab
effects against BACE1 to treat alzheimer’s disease.
Frontiers in Computational Neuroscience, 12(June), 1–
11. https://doi.org/10.3389/fncom.2018.00034
Hu, Q., Feng, M., Lai, L., & Pei, J. (2018). Prediction of
Drug-Likeness Using Deep Autoencoder Neural
Networks. Frontiers in Genetics, 9(November), 1–8.
https://doi.org/10.3389/fgene.2018.00585
Kiriiri, G. K., Njogu, P. M., & Mwangi, A. N. (2020).
Exploring different approaches to improve the success
of drug discovery and development projects: a review.
Future Journal of Pharmaceutical Sciences, 6(1), 27.
https://doi.org/10.1186/s43094-020-00047-9
Li, H., Leung, K. S., Wong, M. H., & Ballester, P. J. (2016).
USR-VS: a web server for large-scale prospective
virtual screening using ultrafast shape recognition
techniques. Nucleic Acids Research, 44(W1), W436–
W441. https://doi.org/10.1093/nar/gkw320
Lipinski, C. A. (2000). Drug-like properties and the causes
of poor solubility and poor permeability. Journal of
Pharmacological and Toxicological Methods,
44(1),
235–249. https://doi.org/10.1016/S1056-8719(00)0010
7-6
Lipinski, C. A. (2004). Lead- and drug-like compounds:
The rule-of-five revolution. Drug Discovery Today:
Technologies, 1(4), 337–341.
https://doi.org/10.1016/j.ddtec.2004.11.007
Liu, R., Sun, Y., Chai, Y., Li, S., Li, S., Wang, L., Su, J.,
Yu, S., Yan, J., Gao, F., Zhang, G., Qiu, H.-J., Gao, G.
F., Qi, J., & Wang, H. (2020). The structural basis of
African swine fever virus pA104R binding to DNA and
its inhibition by stilbene derivatives. Proceedings of the
National Academy of Sciences of the United States of
America, 117(20), 11000–11009.
https://doi.org/10.1073/pnas.1922523117
Maia, E. H. B., Assis, L. C., de Oliveira, T. A., da Silva, A.
M., & Taranto, A. G. (2020). Structure-Based Virtual
Screening: From Classical to Artificial Intelligence.
Frontiers in Chemistry, 8(April).
https://doi.org/10.3389/fchem.2020.00343
Matsumoto, T., Kaifuchi, N., Mizuhara, Y., Warabi, E., &
Watanabe, J. (2018). Use of a Caco-2 permeability
assay to evaluate the effects of several Kampo
medicines on the drug transporter P-glycoprotein.
Journal of Natural Medicines, 72(4), 897–904.
https://doi.org/10.1007/s11418-018-1222-x
Mohs, R. C., & Greig, N. H. (2017). Drug discovery and
development: Role of basic biological research.
Alzheimer’s & Dementia (New York, N. Y.), 3(4), 651–
657. https://doi.org/10.1016/j.trci.2017.10.005
Mortelmans, K., Mortelmans, K., & Zeiger, E. (2016). The
Ames Salmonella / microsome mutagenicity assay The
Ames Salmonella / microsome mutagenicity assay.
5107(December 2000), 29–60.
Mouchlis, V. D., Afantitis, A., Serra, A., Fratello, M.,
Papadiamantis, A. G., Aidinis, V., Lynch, I., Greco, D.,
& Melagraki, G. (2021). Advances in de novo drug
design: From conventional to machine learning
methods. International Journal of Molecular Sciences,
22(4), 1–22. https://doi.org/10.3390/ijms22041676
Muegge, I., Heald, S. L., & Brittelli, D. (2001). Simple
selection criteria for drug-like chemical matter. Journal
of Medicinal Chemistry, 44(12), 1841–1846.
https://doi.org/10.1021/jm015507e
Niel, N., Rechencq, E., Vidal, J. P., Escale, R., Durand, T.,
Girard, J. P., Rossi, J. C., Muller, A., & Bonne, C.
(1992). Synthesis and contractile activity of new
pseudopeptido and thioaromatic analogues of
leukotriene D4. Prostaglandins, 43(1), 45–54.
https://doi.org/10.1016/0090-6980(92)90063-Y
Osborne, D. W., & Musakhanian, J. (2018). Skin
Penetration and Permeation Properties of
Transcutol®—Neat or Diluted Mixtures. AAPS
PharmSciTech, 19(8), 3512–3533.
https://doi.org/10.1208/s12249-018-1196-8
Pen, G., Yang, N., Teng, D., Mao, R., Hao, Y., & Wang, J.
(2020). A review on the use of antimicrobial peptides
to combat porcine viruses.
Antibiotics, 9(11), 1–18.
https://doi.org/10.3390/antibiotics9110801
Petit, J., Meurice, N., Kaiser, C., & Maggiora, G. (2012).
Softening the Rule of Five - Where to draw the line?
Bioorganic and Medicinal Chemistry, 20(18), 5343–
5351. https://doi.org/10.1016/j.bmc.2011.11.064
In Silico De Novo Synthesis, Screening, and ADME/T Profiling of DNA-pA104R Inhibitors as Potential African Swine Fever Therapeutics
25

Pires, D. E. V., Blundell, T. L., & Ascher, D. B. (2015).
pkCSM: Predicting small-molecule pharmacokinetic
and toxicity properties using graph-based signatures.
Journal of Medicinal Chemistry, 58(9), 4066–4072.
https://doi.org/10.1021/acs.jmedchem.5b00104
Polanski, J. (2020). 4.26 - Chemoinformatics☆. In S.
Brown, R. Tauler, & B. Walczak (Eds.),
Comprehensive Chemometrics (Second Edition) (pp.
635–676). Elsevier. https://doi.org/https://doi.org/10.
1016/B978-0-12-409547-2.14327-6
Polgar, T., & M. Keseru, G. (2011). Integration of Virtual
and High Throughput Screening in Lead Discovery
Settings. Combinatorial Chemistry & High Throughput
Screening, 14(10), 889–897.
https://doi.org/10.2174/138620711797537148
Sanchez, G. (2013). Las instituciones de ciencia y
tecnología en los procesos de aprendizaje de la
producción agroalimentaria en Argentina. El Sistema
Argentino de Innovación: Instituciones, Empresas y
Redes. El Desafío de La Creación y Apropiación de
Conocimiento., 26(February), 15–26. https://doi.org/
10.1002/prot
Santos, K. B., Guedes, I. A., Karl, A. L. M., & Dardenne,
L. E. (2020). Highly Flexible Ligand Docking:
Benchmarking of the DockThor Program on the
LEADS-PEP Protein-Peptide Data Set. Journal of
Chemical Information and Modeling, 60(2), 667–683.
https://doi.org/10.1021/acs.jcim.9b00905
Schreyer, A. M., & Blundell, T. (2012). USRCAT: Real-
time ultrafast shape recognition with pharmacophoric
constraints. Journal of Cheminformatics, 4(11), 1.
https://doi.org/10.1186/1758-2946-4-27
Schreyer, A. M., & Blundell, T. L. (2013). CREDO: A
structural interactomics database for drug discovery.
Database, 2013, 1–9.
https://doi.org/10.1093/database/bat049
Shen, M., Tian, S., Li, Y., Li, Q., Xu, X., Wang, J., & Hou,
T. (2012). Drug-likeness analysis of traditional Chinese
medicines: 1. property distributions of drug-like
compounds, non-drug-like compounds and natural
compounds from traditional Chinese medicines.
Journal of Cheminformatics, 4(1), 1–13.
https://doi.org/10.1186/1758-2946-4-31
Singh, N., Chaput, L., & Villoutreix, B. O. (2021). Virtual
screening web servers: designing chemical probes and
drug candidates in the cyberspace. Briefings in
Bioinformatics, 22(2), 1790–1818. https://doi.org/10.
1093/bib/bbaa034
Swinger, K. K., & Rice, P. A. (2004). IHF and HU: Flexible
architects of bent DNA. Current Opinion in Structural
Biology, 14(1), 28–35. https://doi.org/10.1016/j.sbi.20
03.12.003
Umashankar, V., & Gurunathan, S. (2015). Drug discovery:
An appraisal. International Journal of Pharmacy and
Pharmaceutical Sciences, 7(4), 59–66.
Urbano, A. C., & Ferreira, F. (2020). Role of the dna-
binding protein pa104r in asfv genome packaging and
as a novel target for vaccine and drug development.
Vaccines, 8(4), 1–17. https://doi.org/10.3390/vaccines
8040585
Veber, D. F., Johnson, S. R., Cheng, H. Y., Smith, B. R.,
Ward, K. W., & Kopple, K. D. (2002). Molecular
properties that influence the oral bioavailability of drug
candidates. Journal of Medicinal Chemistry, 45(12),
2615–2623. https://doi.org/10.1021/jm020017n
Venkatesh, S., & Lipper, R. A. (2000). Role of the
development scientist in compound lead selection and
optimization. Journal of Pharmaceutical Sciences,
89(2), 145–154. https://doi.org/10.1002/(SICI)1520-
6017(200002)89:2<145::AID-JPS2>3.0.CO;2-6
Wang, J. Y. J. (2001). DNA damage and apoptosis. Cell
Death and Differentiation, 8(11), 1047–1048.
https://doi.org/10.1038/sj.cdd.4400938
Wang, N. N., Dong, J., Deng, Y. H., Zhu, M. F., Wen, M.,
Yao, Z. J., Lu, A. P., Wang, J. B., & Cao, D. S. (2016).
ADME Properties Evaluation in Drug Discovery:
Prediction of Caco-2 Cell Permeability Using a
Combination of NSGA-II and Boosting. Journal of
Chemical Information and Modeling, 56(4), 763–773.
https://doi.org/10.1021/acs.jcim.5b00642
Wang, N. N., Huang, C., Dong, J., Yao, Z. J., Zhu, M. F.,
Deng, Z. K., Lv, B., Lu, A. P., Chen, A. F., & Cao, D.
S. (2017). Predicting human intestinal absorption with
modified random forest approach: a comprehensive
evaluation of molecular representation, unbalanced
data, and applicability domain issues. RSC Advances,
7(31), 19007–19018. https://doi.org/10.1039/C6RA2
8442F
Yates, J. W. T., & Arundel, P. A. (2008). On the volume of
distribution at steady state and its relationship with two-
compartmental models. Journal of Pharmaceutical
Sciences, 97(1), 111–122. https://doi.org/10.1002/jp
s.21089
Zhao, Y. H., Le, J., Abraham, M. H., Hersey, A.,
Eddershaw, P. J., Luscombe, C. N., Boutina, D., Beck,
G., Sherborne, B., Cooper, I., & Platts, J. A. (2001).
Evaluation of human intestinal absorption data and
subsequent derivation of a quantitative structure -
Activity relationship (QSAR) with the Abraham
descriptors. Journal of Pharmaceutical Sciences, 90(6),
749–784. https://doi.org/10.1002/jps.1031
Zhu, Z., Fan, Y., Cai, Z., Zhang, Z., Lu, C., Jiang, T.,
Zhang, G., & Peng, Y. (2019). Prediction of antiviral
drugs against African Swine Fever Viruses based on
protein-protein interaction analysis. BioRxiv.
https://doi.org/10.1101/599043
Zhu, Z., Fan, Y., Liu, Y., Jiang, T., Cao, Y., & Peng, Y.
(2020). Prediction of antiviral drugs against African
swine fever viruses based on protein-protein interaction
analysis. PeerJ, 2020(4), 1–19. https://doi.org/10.
7717/peerj.8855
BIOINFORMATICS 2022 - 13th International Conference on Bioinformatics Models, Methods and Algorithms
26
