
Coordinate Systems for Pangenome Graphs based on the Level Function
and Minimum Path Covers
Thomas B
¨
uchler, Caroline R
¨
ather, Pascal Weber and Enno Ohlebusch
Institute of Theoretical Computer Science, Ulm University, 89069 Ulm, Germany
Keywords:
Pangenome, Coordinate System, Directed Acyclic Graph, Level Function, Minimum Path Cover.
Abstract:
The Computational Pan-Genomics Consortium (Consortium, 2016) described the role of coordinate systems
in genomics as follows: “A pan-genome defines the space in which (pan-)genomic analyses take place. It
should provide a ‘coordinate system’ to unambiguously identify genetic loci and (potentially nested) genetic
variants.” The most natural representations of pangenomes are graphs. The Computational Pan-Genomics
Consortium identified desirable properties of the linear reference genome model that graphical frameworks
should attempt to preserve: spatiality, monotonicity, and readability. In this paper, we introduce a coordinate
system for DAGs that has these properties. It is based on the level function and a minimum path cover of the
graph. Moreover, we describe a new method for finding a minimum path cover in a DAG, which works very
well in practice.
1 INTRODUCTION
Since de novo assembly of mammalian and plant
genomes is still a serious problem, a reference-based
approach is usually used to assemble the genomes of
several individuals of the same species. In this ap-
proach, a high quality genome assembly of a single
selected individual (or a mixture of several individu-
als) is produced, and this assembly is used as a ref-
erence for genomes of the same species. Such a lin-
ear reference provides a coordinate system: the loca-
tion of a genetic element is defined by its coordinate
(starting position) in the reference genome. In rese-
quencing experiments, a high-throughput sequencer
produces DNA-fragments (called reads) of a certain
length (which depends on the technology used) and
each read is then mapped to the locus in the refer-
ence genome at which it fits best. A read can be
mapped correctly if it is similar enough to a subse-
quence of the reference genome. However, since the
reference genome does not represent all known vari-
ations, read mapping tends to be biased towards the
reference and mapping errors may thus occur. For that
reason, reference genomes are more and more being
replaced by pangenomes, which contain the whole ge-
nomic content of a certain species or population. The
genomes of individuals of the same species are usu-
ally very similar. Hence a pangenome often consists
of a reference genome (in form of a DNA sequence
for each chromosome) and a catalog of variations. A
prime example is the human species. The first draft of
the human reference genome, in which known vari-
able regions were poorly represented, was released
in 2001 (Consortium, 2001). To produce a catalog
of all variations in the human population, the 1000
Genomes Project (The 1000 Genomes Project Con-
sortium, 2015) started in 2008. Its original goal was to
sequence the genomes of at least 1000 humans from
all over the world. From the 2504 individuals charac-
terized by the 1000 Genomes Project, it is estimated
that the average diploid human genome has around
4.1 – 5 million point variants such as single nucleotide
polymorphisms (SNPs), multi-nucleotide polymor-
phisms (MNPs), and short insertions or deletions (in-
dels). Moreover, it carries between 2100 and 2500
larger structural variants (SVs) such as large deletions
(or, to a lesser extent, insertions), duplications, copy-
number variation, inversions, and translocations (The
1000 Genomes Project Consortium, 2015). It is quite
natural to model a pangenome as a graph, in which
subsequences that are shared by several individuals
are represented by the same path. The Computational
Pan-Genomics Consortium (Consortium, 2016) de-
scribed the role of coordinate systems in genomics as
follows: “A pan-genome defines the space in which
(pan-)genomic analyses take place. It should provide
a ‘coordinate system’ to unambiguously identify ge-
netic loci and (potentially nested) genetic variants.” If
a pangenome representation is constructed from a ref-
Büchler, T., Räther, C., Weber, P. and Ohlebusch, E.
Coordinate Systems for Pangenome Graphs based on the Level Function and Minimum Path Covers.
DOI: 10.5220/0010165400210029
In Proceedings of the 14th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2021) - Volume 3: BIOINFORMATICS, pages 21-29
ISBN: 978-989-758-490-9
Copyright
c
2021 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
21
erence genome and its variants, the reference genome
can still be used as a coordinate system provided there
is a strict correspondence between positions in the ref-
erence genome and positions in the graph (Dilthey
et al., 2015). However, a graph representation of a
pangenome contains more information than a refer-
ence genome and one should benefit from a coordi-
nate system based directly on the graph. According to
the Computational Pan-Genomics Consortium (Con-
sortium, 2016) there are desirable properties of the
linear reference genome model that graphical frame-
works should attempt to preserve:
• Spatiality: nucleotides that are physically close
together within a genome should have similar co-
ordinates.
• Monotonicity: the genome graph coordinates of
successive nucleotides within a genome should be
increasing.
• Readability: coordinates should be compact and
human interpretable.
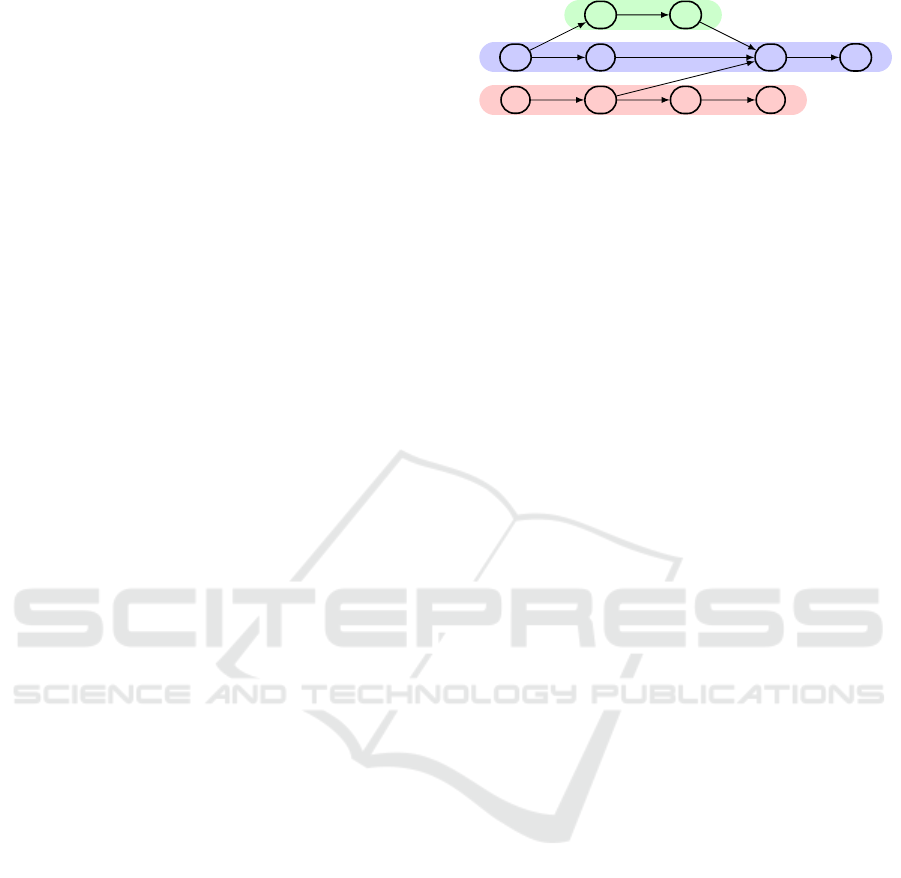
Rand et al. (2017) further distinguish between hori-
zontal and vertical spatiality. Two vertices are hor-
izontally close if their bases are close to each other
in the genome and they are vertically close if they are
variants of each other (in this case, they appear on dif-
ferent paths). For example, the nucleotides G and C
of the sequence TGCG are horizontally close to each
other and the T of the sequence ATAG is vertically
close to the G of the sequence AGGAG; see Fig. 1.
Rand et al. (2017) propose to partition the graph into a
set of non-overlapping sequences, called region paths.
Their approach is still based on a reference genome.
They distinguish two different ways of dividing the
reference genome into region paths:
• Hierarchical partitioning: choose one main region
path through the graph and describe alternative
loci as alternative region paths.
• Sequential partitioning: divide the main region
path whenever an alternative path starts or stops.
Hierarchical partitioning does not fulfill spatiality,
whereas sequential partitioning does. Rand et al.
(2017) also suggested naming schemes and came to
the conclusion that it is difficult to meet all three cri-
teria. The main drawback of their approach is that it
still depends on the existing linear coordinates. It is
still an open problem how to define a non-reference
based coordinate system for a pangenome graph be-
cause graphs admit multiple paths that may have com-
plex relationships. In this paper, we propose a new
kind of coordinate system for directed acyclic graphs
(DAGs). For organisms with linear chromosomes
the restriction to DAGs seems quite natural. Unlike
T G C
A GA T
G G
T
Figure 1: Nucleotide graph that represents the sequences
TGCT, TGAG, ATAG, and AGGAG. The graph has a min-
imum path cover of size 3.
the linear DNA of most eukaryotes, however, typical
prokaryote chromosomes are circular. In this case,
one needs either a linearization of the chromosomes
(for instance by cutting them at the replication ori-
gin) or a “linearization” of the pangenome graph to be
able to apply our method. It should be stressed that a
property like monotonicity can hardly be fulfilled for
cyclic pangenome graphs and therefore a restriction
to non-cyclic graphs seems necessary.
2 BACKGROUND
2.1 Pangenome Graphs
There are several different pangenome represen-
tations, e.g. sets of (un)aligned sequences, nu-
cleotide graphs, de Bruijn graphs, and bidirected
graphs (Consortium, 2016; Paten et al., 2017). A
nucleotide graph is a directed graph G = (V, E)
(where V denotes the set of vertices and E ⊂V ×V the
set of edges), in which walks encode a nucleotide se-
quence (a walk is a sequence of vertices v
1
,v
2
,...,v
k
,
with (v
i
,v
i+1
) ∈ E for 1 ≤ i < k). That is, a nucleotide
graph is a directed graph equipped with a labeling
function V → {A,C, G,T}; see Fig. 1 for an exam-
ple. Here, we assume that every vertex is labeled with
a single nucleotide, but the ideas described in the fol-
lowing can be adapted to graphs with labeling func-
tions that label vertices with nucleotide sequences. In
this paper, we will focus on nucleotide graphs that
are directed acyclic graphs (DAGs). In a DAG, ev-
ery walk is a path: if a vertex v
i
would occur twice in
a walk, then there would be a cycle in the graph. The
length of a path P equals the number of edges of P
and is denoted by |P|.
2.2 Level Function
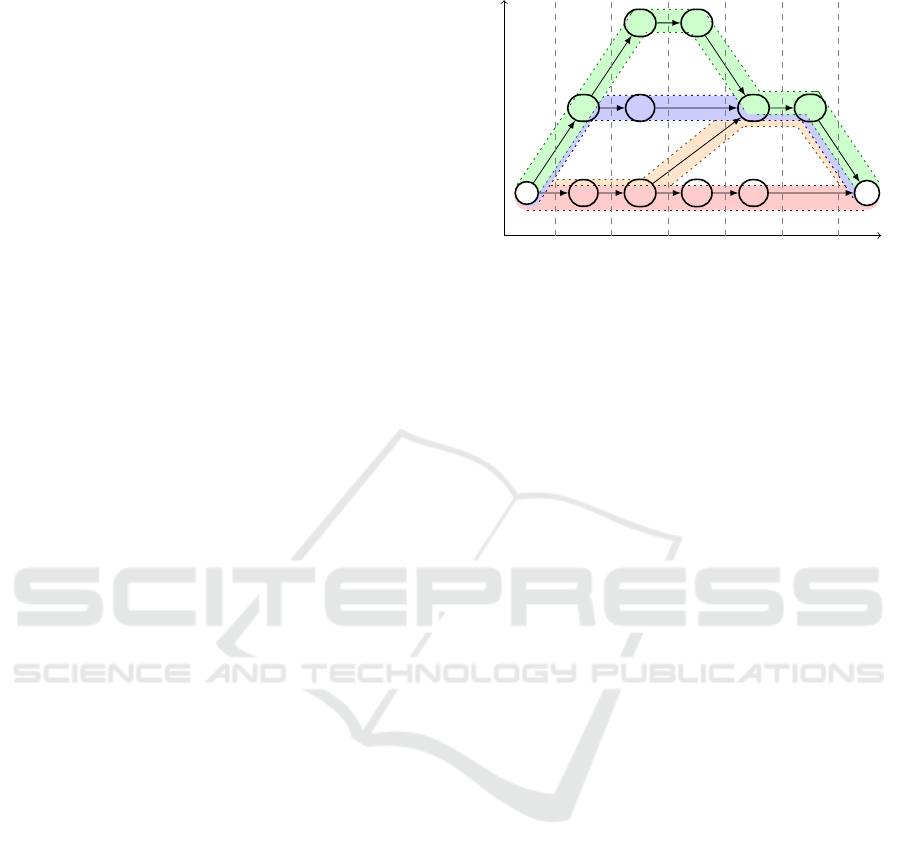
Let G = (V, E) be a DAG with a unique source s ∈ V
(the only vertex with in-degree 0). The function
level : V 7→ N maps a vertex v to its maximum dis-
tance to s (Equation 1); see Fig. 2 for an example.
The level of a vertex can also be defined in a recur-
sive way (Equation 2). The recursive definition yields
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
22
an iterative method to calculate the levels of all ver-
tices, which we will use in our algorithms. For the
rest of the paper, we call level(v) the level of v.
level(v) = max{|P| : P = (s, ...,v) is a path in G}
(1)
level(v) =
(
0 v = s
max({level(u)|(u,v) ∈ E}) + 1 v 6= s
(2)
2.3 Minimum Path Cover Problem
(MPCP)
A path cover PC = {P
1
,..., P
k
} of a DAG G = (V, E) is
a set of paths, so that each vertex v ∈ V is part of a path
P
i
∈ PC. The size of a path cover PC is defined as the
number of paths in PC. Since a single vertex defines a
path, V itself is a path cover of size |V |. The minimum
path cover problem is the problem of finding a path
cover PC of minimum size p
∗
. The minimum size p
∗
is called the width of G.
3 COORDINATE SYSTEM FOR
ACYCLIC NUCLEOTIDE
GRAPHS
To identify a locus in the pangenome, we need a coor-
dinate for each vertex of the nucleotide graph. In this
paper, we propose a new coordinate system for nu-
cleotide graphs that meets the requirements of mono-
tonicity, readability, and (horizontal and vertical) spa-
tiality. The coordinate of a vertex v in our new co-
ordinate system is a pair of two integers. The first
component of the coordinate is the level of vertex v
in the DAG, which measures the distance from the
source to v. The second component is an identifier
of a path to which v belongs. In order to use as few
path identifiers as possible, we will solve the mini-
mum path cover problem. In what follows, we elabo-
rate on this idea and provide an algorithm that gener-
ates such a coordinate system. For technical reasons,
we deal with DAGs that have a unique source s and
a unique sink t (the only vertices with in-degree and
out-degree 0, respectively). Graphs without a unique
source or sink can be modified by adding a super
source s and a super sink t to V , plus edges from s
to all former sources and edges from all former sinks
to t; see Fig. 2. In a graph with a unique source and
sink every vertex lies on some path from the source
to the sink and every path can be extended to a path
from s to t. Hence, there is always a minimum path
P
4
P
1
P
2
P
3
s
T G C
A GA T
G G
T
t
level
0
1 2
3
4
5 6
path
1
2
3
(0,1) (1,1) (2,1) (3,1)
(4,2) (5,2)(1,2) (2,2)
(2,3) (3,3)
(4,1) (6,1)
Figure 2: Nucleotide graph with super source and sink,
which represents the sequences TGCT, TGAG, ATAG and
AGGAG. The set {P
1
,P
2
,P
3
,P
4
} is not a minimum path
cover, but {P
1
,P
2
,P
3
} is. The paths in this figure all start
at s and end at t.
cover that solely consists of paths from s to t. If ` is
the length of the longest path in the nucleotide graph
G and PC
∗
= {P
1
,..., P
p
∗
} is a minimum path cover
of G (w.l.o.g. all paths in PC
∗
start at s and end at t),
then the coordinates of s and t will be (0, 1) and (`,1)
respectively and the coordinates of all other vertices
are elements of {1, ..., ` − 1} × {1, ..., p
∗
}. For a ver-
tex v ∈ V and a path cover PC, we denote the set of
all paths that include v by PC(v). One of the paths in
PC(v) is chosen to be the representative of v. A possi-
ble choice is the path with lowest id, hence the coordi-
nate of a vertex v is (level(v), min(PC
∗
(v))). Figure 2
depicts a DAG with a path cover and the coordinates
of its vertices.
The two coordinates can be thought of as a hor-
izontal and a vertical coordinate. The horizontal
coordinate—similar to the coordinate in a linear ref-
erence genome—is the offset to the start of the chro-
mosomal DNA sequences. The vertical coordinate is
a path identifier, which can be chosen in such a way
that it carries useful information. For example, one
could assign the vertices of the most likely genome
(the reference) to the path with id 1, the vertices of
the second most likely haplotype to the path with id
2, etc.
Moreover, this coordinate system satisfies the cri-
teria mentioned above. Having only two coordinates,
which both carry useful information, can be called
’readable’. Monotonicity follows directly from the
use of the level function in the first coordinate: the
genome graph coordinates of successive bases within
a genome are increasing. Finally, the coordinate sys-
tem also meets the requirement ’spatiality’. If two
vertices represent a variant of each other, for instance
a SNP, then they will be at the same level and share
their first coordinate. Thus, we have vertical spatial-
Coordinate Systems for Pangenome Graphs based on the Level Function and Minimum Path Covers
23

ity. Two vertices that represent nucleotides that are
close to each other in the genome will be at levels
close to each other. If they belong to the same haplo-
type, they will be covered by the same path. In this
way, horizontal spatiality is achieved.
To generate such a coordinate system, we calcu-
late the level of each vertex and solve the MPCP. The
coordinate of a vertex can then easily be reported as
a combination of its level and the identifier of a path
that covers it.
4 SOLVING THE MPCP
The classical solution of the MPCP works as follows.
The MPCP is reduced to a maximum cardinality bi-
partite matching problem, which in turn is reduced to
a maximum flow problem; see Cormen et al. (2009).
Since the latter can be solved by the Ford-Fulkerson
method, this method can also solve the MPCP. This
approach takes O (n
3
) time (in this paper we assume
|V | = n and |E| = m).
M
¨
akinen et al. (2015) described a different
method to solve the MPCP. They reduced this prob-
lem first to a minimum cost flow problem, and then
to a minimum cost circulation problem. Overall, their
solution takes O(nm + n
2
log(n)) time.
Kuosmanen et al. (2018) introduced an algo-
rithm that first generates an initial solution by us-
ing a method based on a classical greedy set cover
algorithm and then optimizes that solution with a
flow algorithm. They showed that the initial path
cover has the size p ≤ p
∗
log(n), where p
∗
is the
width of the DAG. Therefore, their algorithm runs in
O(m p
∗
log(n)) time.
We will use a method comparable to the latter.
First, an initial path cover PC of size p will be cal-
culated in O(p m) time using a heuristic algorithm.
Then a network flow algorithm, which makes use of
the Ford-Fulkerson method, is employed to reduce
the path cover to minimum size. A description of
the network flow algorithm can be found in Kuosma-
nen et al. (2018, Section 2) and will not be repeated
here. Minimizing the flow takes O ((p − p
∗
)m) time
because finding and adding a flow requires O (m) time
and p− p
∗
flows will be added. To report a path, O(m)
time is needed. Since there are p
∗
paths, reporting all
paths takes O(p
∗
m) time. In total, our method gener-
ates a minimum path cover in O (pm) time, where p
is the size of the initial path cover.
Algorithm 1: Calculate an initial path cover. An entry
path[i] in the array path is a list of all vertices that are
contained in path i.
Data: DAG G = (V,E) with source node s,
a vector d
in
of size |V | containing the
in-degree of each node
Result: An array level of size |V | containing the
level of each node, a set R
v
for each node
v, which contains all candidate paths for v.
1 level[s] ← 0
2 l ← 0, Q
l
← {s}, Q
l+1
←
/
0
3 p ← 0
4 while Q
l
6=
/
0 do
5 for v ∈ Q
l
do
/* choose a path to cover v */
6 R
v
←
/
0
7 for (u, v) ∈ E do
8 for i ∈ R
u
do
9 last ← last element of path[i]
10 if level[last] < level[u] ∨ last = u
then
11 R
v
← R
v
∪ {i}
12 if R
v
=
/
0 then
/* add a new path */
13 p ← p + 1
14 R
v
← {p}
15 path[p] ← [v]
16 else
/* the path min(R
v
) is chosen
to cover v */
17 i ← min(R
v
)
18 find the path P from the last node of
path i to v
19 append P to path[i]
20 for (v, w) ∈ E do
21 d
in
[w] ← d
in
[w] − 1
22 if d
in
[w] = 0 then
23 level[w] ← l + 1
24 Q
l+1
← Q
l+1
∪ {w}
25 l ← l + 1
26 Q
l
← Q
l+1
27 Q
l+1
←
/
0
4.1 Generating an Initial Path Cover
Algorithm 1 generates an initial path cover. It tra-
verses the graph level-wise from s to t and the path
cover is computed iteratively by extending the cur-
rent partial path cover to the next level. At level 0 the
source vertex s is covered by path 1. While visiting
the vertices of level l, the vertices of level l + 1 are
calculated. Furthermore, the algorithm tries to cover
all vertices of level l by extending paths that end at a
level < l with a vertex of level l. If no suitable path
can be found for a vertex, the algorithm will add a
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
24

new path. In the following, it will be explained how
the level of a vertex is calculated and afterwards how
the path cover is generated.
The array level stores the level of each vertex v,
i.e. level[v] = level(v). The queue Q
l
contains all ver-
tices of level l. Initially l is 0, Q
l
= {s} and level[s] =
0 because the source is the only vertex in level 0. Fur-
thermore, there is an array d
in
, which initially stores
the number of incoming edges for each vertex. When
a vertex v at level l is encountered, d
in
[w] is decre-
mented by one for each successor w of v. If d
in
[w] = 0,
then all predecessors of w have been visited before.
In this case, level(w) = level(v) + 1 = l + 1 because
v has highest level among all the predecessors of w.
Lines 23-24 of Algorithm 1 handle this case by set-
ting level[w] = l + 1 and adding w to the queue Q
l+1
.
When all vertices of level l have been dealt with, the
algorithm will start the next iteration with the vertices
of the level l + 1 (lines 25-27). With this method, the
vertices are visited level by level. We now explain
how the algorithm covers the vertices with paths.
The counter p stores the number of paths used so
far (upon termination, p is the size of the path cover)
and for each path identifier i, 1 ≤ i ≤ p, there is a
list of vertices path[i] in the array path. In the first
iteration, the algorithm will add the path path[1] = [s].
This means that the first path consists of the vertex s.
Moreover, the algorithm calculates a set R
v
for
each vertex v, which contains the identifiers of all
candidate paths for v, i.e. all paths that can poten-
tially cover v. A path that covers v must be selected
from this set R
v
. In the following, we choose min(R
v
)
to be that path. A condition for a path i to be in
R
v
is that i was a candidate for a predecessor u of v
i.e. i ∈ R
u
. It follows as a consequence that R
v
is a
subset of
S
(u,v)∈E
R
u
, but in general the sets are not
equal. For example, a path i ∈
S
(u,v)∈E
R
u
might have
already been used to cover a vertex w 6= v at level
l = level(v). We use a case distinction to find the can-
didate paths. Let i ∈ R
u
, where u is a predecessor of
v, and let last be the last element of path[i]. If (a)
level[last] < level[u], then i was a candidate path for
u that was neither chosen to cover u nor one of its suc-
cessors. Hence it is a candidate path for v. If (b) path i
ends at u (i.e. last = u), then i is also a candidate path
for v.
Algorithm 1 calculates R
v
by the case distinction
described above (lines 8-11). If R
v
is empty after this
step, none of the existing paths can be used to cover
v. In this case, the algorithm adds a new path with
identifier i
new
= p + 1 and uses this to cover v (lines
13-15). Otherwise, if R
v
is non-empty after this step,
the path with identifier i = min(R
v
) is chosen to cover
v and path[i] is updated accordingly (lines 17-19). To
this end, one must find a path P (implemented as a list
of vertices) from the last vertex last of path i to v. This
is done be tracing a path from v back to last. Initially,
the list P just contains v. Then vertices are added at
the front of the list until the vertex last is reached.
A vertex u can extend the path P at the front if (a)
it is a predecessor of the current front, (b) i ∈ R
u
, and
(c) level[u] > level[last]. If we repeat this process, we
will ultimately reach last because the condition at line
10 of Algorithm 1 ensures that, among all vertices at
level level(last), only last can propagate the path i.
Upon termination of Algorithm 1, the paths in ar-
ray path form a path cover. However, we are solely
interested in paths from s to t. In what follows we
explain Algorithm 2, that extends the paths on both
ends. Let v
1
and v
2
be the first and the last vertex of
path[i], respectively. We have to add a subpath from s
to a predecessor of v
1
and a subpath from a successor
of v
2
to t to obtain a path from s to t. We can find
such subpaths by a depth first search in linear time, or
we can copy them from other paths. Note that path
1 is a path from s to t already, since 1 is the smallest
path identifier and will therefore be used continuously
from vertex s to t. For i 6= 1 all predecessors of v
1
are covered by paths < i because v
1
is the first vertex
covered by a path ≥ i. Likewise, all successors of v
2
are covered by paths < i because i was not chosen to
cover one of them. These observations lead to an it-
erative process for finding the subpaths that shall be
copied. When extending path i, all paths < i are paths
from s to t. Among all predecessors u of v
1
, we select
the one that is covered by the path with the smallest
identifier j. Since j < i, j is a path from s to t and we
can copy the subpath from s to u. In the same way, we
can find a path from a successor of v
2
to t. Algorithm
2 extends each path to be a path from s to t.
Algorithms 1 and 2 compute an initial path cover
in O(p m) time, where p is the size of the cover and
m is the number of edges in the DAG. This is because
every edge is considered exactly once in line 7 of Al-
gorithm 1 and the loop in line 8 of Algorithm 1 is
iterated at most p times. Moreover, the overall time
required by line 18 of Algorithm 1 and Algorithm 2
is also O(p m) because for each of the p paths each
edge is considered at most once. (If one wants to cal-
culate the minimum path cover as described above,
one could directly insert flow into the flow network
instead of storing and reporting a path.)
4.2 A Simple Heuristic
In general, Algorithms 1 and 2 do not deliver a mini-
mum path cover; see Fig. 3. This is because the order
in which vertices are covered by paths plays a key
Coordinate Systems for Pangenome Graphs based on the Level Function and Minimum Path Covers
25
Algorithm 2: After iteration i, path[i] is a path from s
to t. Since j and k must be smaller than i, these paths
are already paths from s to t.
Data: DAG G = (V, E), path array path and can-
didate sets R
V
after applying Algorithm 1
Result: Path array path contains paths from s to t
1 for i ← 2 to p do
/* path 1 is already a path from s to t
*/
2 j,k ← ∞
3 u
j
,u
k
← ⊥
4 v
1
← first element of path[i]
5 v
2
← last element of path[i]
6 for (u,v
1
) ∈ E do
7 if min(R
u
) < i then
8 u
j
← min(R
u
)
9 u
j
← u
10 for (v
2
,u) ∈ E do
11 if min(R
u
) < k then
12 k ← min(R
u
)
13 u
k
← u
14 Prepend the vertices of the subpath from s to u
j
of path[ j] to path[i]
15 Append the vertices of the subpath from u
k
to t
of path[k] to path[i]
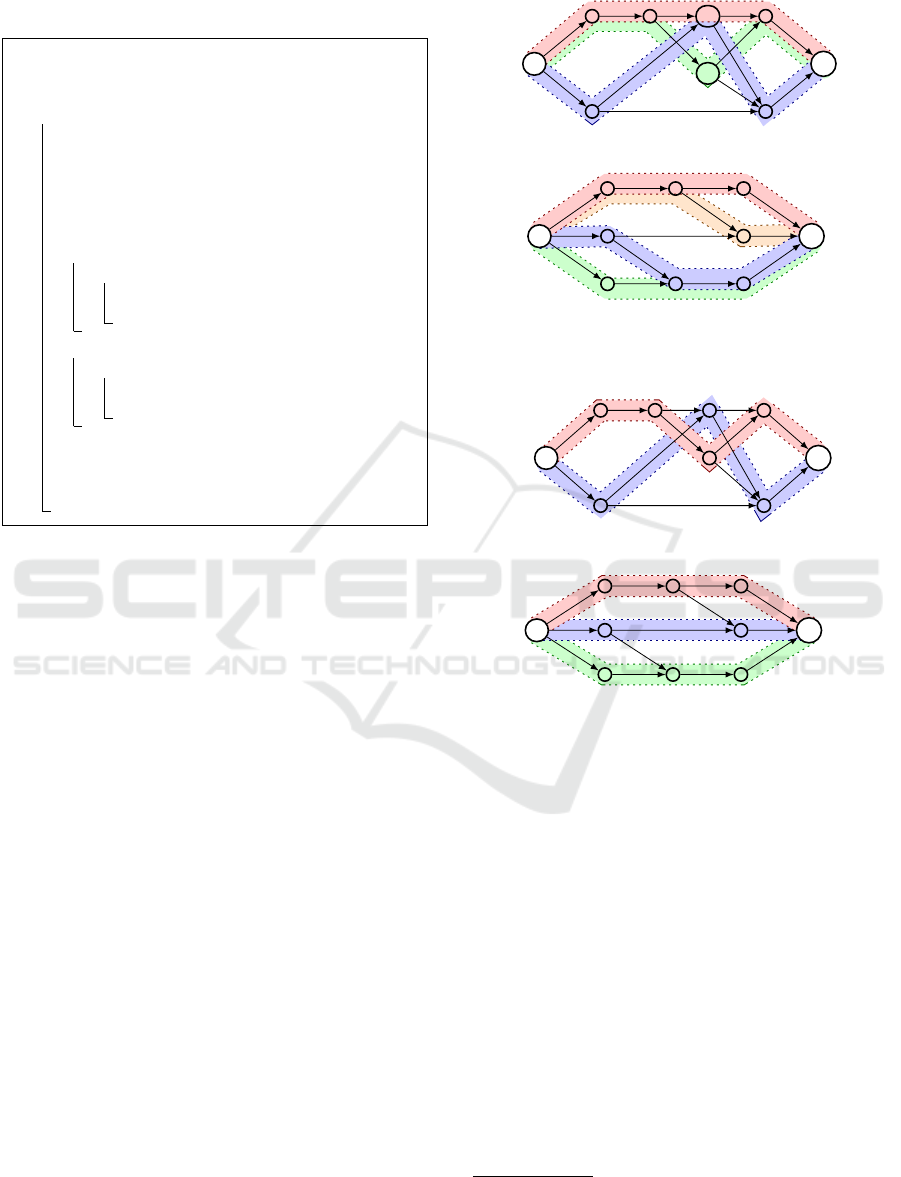
role. For example, in level 3 of the graph depicted
in Fig. 3.a, the vertices u and v must be covered with
paths. The candidate paths for vertex u are 1 and 2,
whereas only path 1 reaches vertex v. Algorithm 1
will first cover vertex u with path 1 because this is the
smallest candidate (i.e. min(R
u
) = 1). It follows that
vertex v cannot be covered by path 1 and consequently
a new path 3 must be added to cover this vertex. A
better solution would be to cover vertex u with path
2 and vertex v with path 1, as shown in Fig. 3.c. It is
possible to improve the size of an initial path cover (in
many cases) by modifying the way in which a path is
chosen to cover a vertex (i.e. it is not necessarily the
path with identifier i = min(R
v
)).
To get a locally optimal solution (i.e. an optimal
solution for a fixed level l), we could solve a maxi-
mum cardinality bipartite matching problem. Define
a bipartite graph as follows. Let X be the set of all
vertices in Q
l
and let Y be the set Y =
S
v∈X
R
v
. Fur-
thermore, there is an edge between v ∈ X and i ∈ Y
if and only if i ∈ R
v
. Now the problem is to find a
maximum cardinality matching in the bipartite graph,
i.e. a matching that contains the largest possible num-
ber of edges. However, the example in Fig. 3.b and
3.d shows that the locally optimal solutions do not
yield a globally optimal solution. This is because one
of the two solutions to the maximum cardinality bi-
partite matching problem at level 2 in Fig. 3.b would
cover the upper vertex with path 1 (the only possi-
bility) and the lower vertex with path 2. In level 3,
Calculated path cover:
a)
s
u
v
t
{1}
{1}
{2}
{1} {1,2}
{3}
{1,3}
{2,3}
{1,2,3}
b)
s
t
{1}
{1}
{2}
{3}
{1}
{2,3}
{1}
{4}
{2,3}
{1,2,3,4}
Minimum path cover:
c)
s
t
{1}
{1}
{2}
{1} {2}
{1}
{1}
{2}
{1,2}
d)
s
t
{1}
{1}
{2}
{3}
{1}
{3}
{1}
{2}
{3}
{1,2,3}
Figure 3: Different examples of path covers. For each ver-
tex v, the set R
v
is shown below v. The path covers depicted
in a) and b) are calculated by Algorithms 1 and 2, whereas
c) and d) show the corresponding minimum path covers.
however, it becomes clear that a globally optimal so-
lution (a minimum path cover) can only be obtained
if the lower vertex at level 2 is covered with path 3.
Since the computation of locally optimal solutions
is time-consuming and does not yield a globally op-
timal solution anyway, we will use a simple heuristic
to obtain local solutions. In each level l, we use a
bucket sort to sort the vertices v in Q
l
in increasing
order by the number of candidate paths (the cardinal-
ity of R
v
). Then we process the vertices v in that order
as follows:
1. If R
v
is empty, assign a new path to v and continue
with the next vertex.
2. Otherwise, take the first element i from R
v
.
1
1
In our implementation R
v
is implemented as an ordered
set and the elements are processed in that order.
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
26

• If i was already used to cover another vertex at
the current level, remove i from R
v
and continue
at (1).
• Otherwise, assign i to v and continue with the
next vertex.
The intuition behind this procedure is simple. For a
singleton set R
v
= {i}, there is no choice: we must
assign i to v. Once we have done this, i cannot cover
another vertex from Q
l
, but a vertex u with a big can-
didate set R
u
has more options than v. Thus, if we
start with small candidate sets, there is a fair chance
that a big candidate set R
u
still contains a path that can
be assigned to u when it is u’s turn.
5 EXPERIMENTAL RESULTS
We implemented the algorithms described above and
built a prototype, that generates a coordinate sys-
tem for a DAG. The software and scripts to replicate
the experiments are available at: www.uni-ulm.de/
in/theo/research/seqana. The program takes a graph
in GFA format as input, computes a path cover and
the corresponding coordinate system, and outputs the
graph (in which vertices are aligned with their coor-
dinates) in DOT format.
Three different strategies were used to compute an
initial path cover:
• the greedy set cover method (gsc) proposed by
Kuosmanen et al. (2018),
• our level-wise computation of the cover (lev), and
• our simple heuristic (sh).
The calculation of the minimum path cover was tested
with real world data and generated data. All experi-
ments were conducted on a Ubuntu 16.04.4 LTS sys-
tem with two 16-core Intel
R
Xeon
R
E5-2698 v3 pro-
cessors and 256 GB RAM.
We used the variation graph toolkit vg (Gar-
rison et al., 2018) to construct a graph from the
FASTA
2
and VCF
3
files for the first human chromo-
some provided by the 1000 genome project (The 1000
Genomes Project Consortium, 2015). In vg, vertices
are not labeled with single nucleotides (as required
here), but with nucleotide sequences. That is why our
program replaces each vertex in the vg graph by a path
of vertices with nucleotides as labels. The resulting
graph has more than 255 ×10
6
vertices and 262 ×10
6
2
Available at: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp
/technical/reference/phase2 reference assembly sequence/
3
Available at: ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp
/release/20130502/
edges. Since the graph has a relatively simple struc-
ture, all three strategies deliver an initial path cover
of size 9 and the initial path cover is a minimum path
cover already. Using the gsc strategy, the overall cal-
culation of the coordinates takes 48 min. The other
strategies lev and sh require 64 and 94 min, respec-
tively.
To get more complex graphs, we generated graphs
in two different ways. The first method is inspired
by the well-known Erdos-Renyi model (Erd
˝
os and
R
´
enyi, 1959) in which a graph is chosen uniformly
at random from the collection of all graphs which
have n vertices and m edges. We modified the model
in such a way that acyclic graphs with a unique
source and sink are generated. The set of vertices
is V = {1, . . . ,n} and the set of edges E initially is
{(1,u), (u, n)|u ∈ {2, . .. , n − 1}}. This ensures that 1
is the unique source and n is the unique sink. Then
two vertices u and v are chosen uniformly at random
from V \ {1, n}. If u < v then (u, v) is added to E, if
v < u then (v, u) is added to E, and if u = v no edge
is added to E. This process is repeated until the graph
contains m edges.
Our second method tries to generate pangenome
graphs that are more complex than the currently avail-
able pangenome graphs (as the one described above).
The parameters of this model are the number n of ver-
tices, a maximum size p for a path cover, and a path
length `. Again, V is {1, . . ., n}. The set of edges
E is a union of the edges of p generated paths. The
first p − 1 paths are generated by choosing `+1 (pair-
wise different) vertices from {2,... , n − 1} uniformly
at random.
The ` + 1 vertices are then sorted in increasing or-
der, say u
1
< · ·· < u
`+1
. Finally, the edges (1,u
1
),
(u
i
,u
i+1
) for 1 ≤ i ≤ `, and (u
`+1
,n) are added to E.
The last path initially contains all vertices of V that
are not part of any of the previous paths. If this path
smaller than `, randomly chosen vertices are added
to the path until it has the length ` (depending on the
parameter, the last path can be significant longer than
`). This generates a DAG with the unique source 1
and the unique sink n. Furthermore, it is clear that the
width of the graph is at most p.
We generated 200 graphs with each model. In the
Erdos-Renyi model, we used n = 10
6
vertices and be-
tween 2 × 10
6
and 4 × 10
6
edges. The number of lev-
els in the generated graphs varied between 420 and
830. In our second model, the parameter for the num-
ber of vertices was also n = 10
6
. The number of paths
was varied between 100 and 200 and the path length
was varied between 5,000 and 10,000. The graphs
generated in this way have between 1×10
6
and 2×10
6
edges and between 150,000 and 600,000 levels.
Coordinate Systems for Pangenome Graphs based on the Level Function and Minimum Path Covers
27
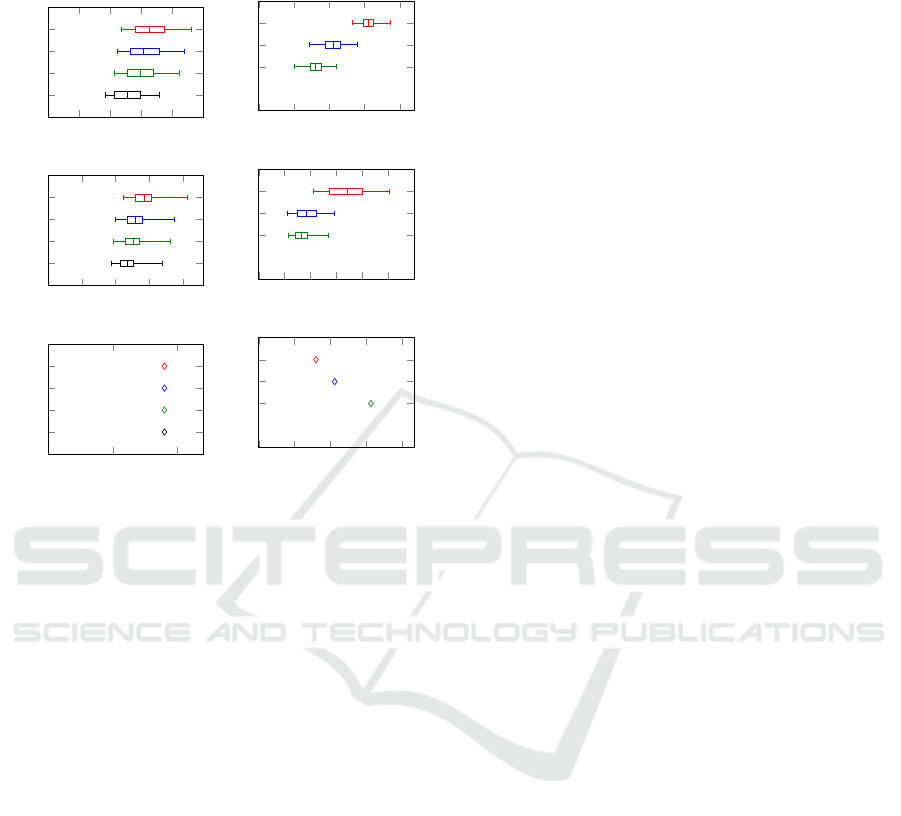
Size of initial path cover Time for generating
the coordinate system
0 100 200
p∗
p
sh
p
lev
p
gsc
s
0
50
100
150
200
t
sh
t
lev
t
gsc
sec.
0 100 200
p∗
p
sh
p
lev
p
gsc
s
0 100 200
t
sh
t
lev
t
gsc
sec.
0 10
p∗
p
sh
p
lev
p
gsc
min
0 30
60
90 120
t
sh
t
lev
t
gsc
min.
Figure 4: Comparison of the different approaches: the
greedy set cover (gsc), our level-wise computation of the
cover (lev), and our simple heuristic (sh). On the top:
graphs generated by the Erdos-Renyi model. In the middle:
graphs generated by the second model. On the bottom: the
graph generated from real world data using the vg toolkit.
Recall that p
∗
denotes the width of the graph (the size of a
minimum path cover).
In our experiments, we measured the size of the
initial solution and the time to calculate the coordi-
nate system (this includes the time for flow minimiza-
tion). The distributions of the measured values are
shown in Figure 4. It can be seen that our method
calculates smaller initial path covers than the greedy
set cover strategy and that the simple heuristic further
improves the result. To be more precise, in each of
the 400 test cases, the initial path cover constructed
by our strategy was smaller compared to that gen-
erated by greedy set cover strategy. Furthermore, in
98% of the test cases the simple heuristic sh returned
a smaller initial path cover than the lev method. We
would like to stress that the overall run-time strongly
depends on the time required for flow minimization,
i.e. the smaller the initial path cover the better. That
is why the simple heuristic outperforms the other ap-
proaches in most cases. However, the cost of using the
simple heuristic cannot be compensated by the faster
flow minimization in all cases.
6 DISCUSSION
In this paper, we proposed a new coordinate system
for acyclic nucleotide graphs, which meets the re-
quirements monotonicity, readability as well as hor-
izontal and vertical spatiality. The first part of the
coordinate is the level of a vertex, which in essence
is the offset from the start of the genomic sequence
(chromosome). The second part is a path identifier,
which can be interpreted as a vertical coordinate. By
using a minimum path cover, the number of path iden-
tifiers is reduced to a minimum, which leads to more
comprehensible coordinates.
The minimum path cover can be calculated by an
algorithm that first finds an initial solution and then
transforms this solution into a minimum path cover.
The run-time of the algorithm strongly depends on
the quality of the initial solution. Our algorithm sh
generates the smallest initial solutions. The lev strat-
egy is the fastest in finding an initial solution in the
generated graphs. In graphs with low width p
∗
, such
as the graph constructed by vg using a FASTA and
VCF file, the gsc strategy is faster. Since all strate-
gies are already finding minimum solutions in such
graphs, the initialization, instead of the minimization,
is the crucial step. Therefore, using the gsc strategy is
best for the tested real world data. In more complex
graphs, the strategy sh is superior.
It should be investigated how additional informa-
tion like haplotypes or frequency of alleles can be in-
corporated into the second coordinate, because this
would make the coordinate systems even more infor-
mative.
ACKNOWLEDGEMENTS
Funding: This work was supported by the DFG (OH
53/7-1).
REFERENCES
Consortium, I. H. G. S. (2001). Initial sequencing and anal-
ysis of the human genome. Nature, 409:860–921.
Consortium, T. C. P.-G. (2016). Computational pan-
genomics: status, promises and challenges. Briefings
in Bioinformatics, 19(1):118–135.
Cormen, T. H., Leiserson, C. E., Rivest, R. L., and Stein,
C. (2009). Introduction to Algorithms. MIT Press,
Cambridge, MA.
Dilthey, A., Cox, C., Iqbal, Z., Nelson, M. R., and McVean,
G. (2015). Improved genome inference in the mhc
using a population reference graph. Nature Genetics,
47(6):682–688.
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
28

Erd
˝
os, P. and R
´
enyi, A. (1959). On random graphs. Publi-
cationes Mathematicae, 6:290–297.
Garrison, E., Sir
´
en, J., Novak, A. M., Hickey, G., Eizenga,
J. M., Dawson, E. T., Jones, W., Garg, S., Markello,
C., Lin, M. F., et al. (2018). Variation graph toolkit
improves read mapping by representing genetic varia-
tion in the reference. Nature biotechnology.
Kuosmanen, A., Paavilainen, T., Gagie, T., Chikhi, R.,
Tomescu, A., and M
¨
akinen, V. (2018). Using min-
imum path cover to boost dynamic programming on
dags: Co-linear chaining extended. In Raphael, B. J.,
editor, Research in Computational Molecular Biology,
pages 105–121, Cham. Springer International Pub-
lishing.
M
¨
akinen, V., Belazzougui, D., Cunial, F., and Tomescu,
A. I. (2015). Genome-scale algorithm design. Cam-
bridge University Press.
Paten, B., Novak, A. M., Eizenga, J. M., and Garrison, E.
(2017). Genome graphs and the evolution of genome
inference. Genome Research, 27(5):665–676.
Rand, K. D., Grytten, I., Nederbragt, A. J., Storvik, G. O.,
Glad, I. K., and Sandve, G. K. (2017). Coordinates
and intervals in graph-based reference genomes. BMC
Bioinformatics, 18(1):263.
The 1000 Genomes Project Consortium (2015). A global
reference for human genetic variation. Nature,
526:68–74.
Coordinate Systems for Pangenome Graphs based on the Level Function and Minimum Path Covers
29
