
Big Data Scalability of BayesPhylogenies on Harvard’s Ozone 12k Cores
M. Manjunathaiah
1,2
, A. Meade
3
, R. Thavarajan
1
, P. Protopapas
1
and R. Dave
1
1
IACS, SEAS, Harvard University, U.S.A.
2
CS, SMPCS, University of Reading, U.K.
3
SBS, University of Reading, U.K.
Keywords:
Big Data, Phylogenetics, Exascale.
Abstract:
Computational Phylogenetics is classed as a grand challenge data driven problem in the fourth paradigm of
scientific discovery due to the exponential growth in genomic data, the computational challenge and the poten-
tial for vast impact on data driven biosciences. Petascale and Exascale computing offer the prospect of scaling
Phylogenetics to big data levels. However the computational complexity of even approximate Bayesian meth-
ods for phylogenetic inference requires scalable analysis for big data applications. There is limited study on
the scalability characteristics of existing computational models for petascale class massively parallel comput-
ers. In this paper we present strong and weak scaling performance analysis of BayesPhylogenies on Harvard’s
Ozone 12k cores. We perform evaluations on multiple data sizes to infer the scaling complexity and find that
strong scaling techniques along with novel methods for communication reduction are necessary if computa-
tional models are to overcome limitations on emerging complex parallel architectures with multiple levels of
concurrency. The results of this study can guide the design and implementation of scalable MCMC based
computational models for Bayesian inference on emerging petascale and exascale systems.
1 INTRODUCTION
Computational Phylogenetics is classed as a grand
challenge data driven problem in the fourth paradigm
of scientific discovery due to the exponential growth
in genomic data, the computational challenge and
the potential for vast impact on data driven bio-
sciences(Warnow, 2017). Constructing the ”tree of
life” in the orders of 100k taxa and beyond is a big
data computational grand challenge. Such trees offer
insights into evolutionary processes at deep spatio-
temporal scales. Constructing the tree of life with
millions of species each with genomes in the order
of millions of nucleotides will depend on methods,
analysis and computational systems that are radically
scaled to offer new scalable solutions.
Given n species (or taxa) a phylogenetic tree
T (V,E) is a representation of inter-relationship
amongst the taxa using any data: molecular or mor-
phological. The tree T is typically rooted and binary
(bifurcating) with: n ∈ V the leaves, the degree of
internal nodes is 3, except for the root. The phyloge-
netic inference problem is to construct T such that the
labellings of the leaves correspond to the evolutionary
history of the given set of species. The computational
challenge of constructing the tree is apparent from the
number of possible topologies for n species.
(2n − 3)!
2
n−2
(n − 2)!
For a rooted tree, the number of possible topolo-
gies is 8 ∗ 10
21
for just n = 20 taxa. Thus the tractable
approaches are invariably heuristic based involving
some form of a search of the combinatorial search
space of possible trees.
State-of-the-art computational techniques are
rooted in Bayesian Inference following the first for-
mulation that cast the phylogeny as a random variable,
thereby enabling the inference problem to be studied
in well-founded statistical frameworks(Felsenstein,
2004). BayesPhylogenies incorporates a general
analysis framework for inferring phylogenies with
the Metropolis-coupled Markov chain Monte Carlo
(MCMCMC) method(Pagel and Meade, 2006). An
integrated software system, BayesPhy and BayesTrait
has been implemented with the goal of generating sta-
tistically robust results for phylogenetic inference and
comparative analysis. These modelling and analysis
software systems have been made available to the bio-
science research community, available on Harvard’s
Supercomputer Odyssey(RC-FAS, 2018a), adopted
Manjunathaiah, M., Meade, A., Thavarajan, R., Protopapas, P. and Dave, R.
Big Data Scalability of BayesPhylogenies on Harvard’s Ozone 12k Cores.
DOI: 10.5220/0007249601430148
In Proceedings of the 12th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2019), pages 143-148
ISBN: 978-989-758-353-7
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
143

by the scientific community, cited over 3000 times in
the literature.
Phylogenetic inference (PI), constructing evolu-
tionary histories, and Phylogeny Comparative Meth-
ods (PCM) a complementary statistical framework for
analysing data in a phylogenetic context, are funda-
mental and universal methods for studying biological
systems(Pagel and Meade, 2008). With large volumes
of data produced by next generation sequencing large
phylogenies have been created: these include a near
complete phylogeny of the birds (9000 taxa) and a
large fish phylogeny (8000 taxa), these phylogenies
join a near complete mammal phylogeny (5000 taxa),
a mega-phylogeny of plants (55,000+ taxa). PCM
analysis has also been applied to discover many evo-
lutionary processes.
Despite these advances, the Phylogenetic problem
remains far from being considered as ’solved’ and
in fact has become acute at big data scale. It was
hoped that access to large data sets would make phy-
logenetic inference universally available, robust and
accurate, surprisingly the opposite appears to be the
case(Salichos and Rokas, 2013). Analysing big data
is therefore much more than redesigning current mod-
elling used for traditional analysis to work with data
sets orders of magnitude larger. New generations of
models, methods and software are needed to fully ex-
ploit the power of these data sets. Scalability is a
defining feature of big data analysis. Overly simple
statistical models do not scale, software designed to
work with data sets orders of magnitude larger than it
was originally designed are not scalable, and compu-
tational methods to convert the results to biologically
relevant information also have scalability limitations.
These can be defined as model scalability, software
scalability, and data visualisation scalability. Big
data phylogenetic inference and comparative meth-
ods offer much more than the challenge of analysing
larger data sets: they offer new opportunities to exam-
ine evolutionary processes and the prospect of gaining
new insights at both micro and macro levels.
In this paper we present evaluation of scalabil-
ity of Bayesphylogenies across two petascale class
architectures on fish big data data sets (3k and 5k).
Analysis of strong and weak scaling on large proces-
sor counts from 4k to 12k gives insights into hybrid
parallel program efficiencies, scalability limitations
across large taxa sets and algorithmic characteristics
of MCMCMC. From these insights we make sev-
eral conclusions on scaling phylogenetic analysis to
next generation from hierarchical parallelisation and
emerging algorithmic adaptations based on machine
learning techniques.
2 BIG DATA BAYESIAN
PHYLOGENETICS
2.1 Bayesian Phylogenetics
2.1.1 Search Procedure
Algorithmic approaches to Bayesian inference of
phylogenies have two components: a scoring metric
and a search procedure.
Let D denote the DNA sequence data from n taxa.
An analysis is typically performed over 10,000 nu-
cleotides drawn from multiple genes. The phyloge-
netic analysis seeks to infer the tree τ
i
that is most
consistent with the observed data D. As the space of
possible trees is exponential in the number of taxa the
problem is framed as search of plausible trees condi-
tional on the observation in a statistical framework:
P(τ
i
|D). A computational procedure is then formu-
lated using Bayes rule as follows:
P(τ
i
| D) =
P(τ
i
) · P(D/τ
i
)
∑
B(n)
j=1
P(τ
i
) · P(D/τ
i
)
(1)
where P(τ
i
| D) gives a score for the i
th
tree,
P(D/τ
i
) the likelihood and P(τ
i
) the prior probaba-
bility.
Instead of computing one tree as in maximum-
likelihood method the formulation in equation 1 com-
putes a distribution of trees by applying a MCMC
search procedure (Meade, 2011). Under the assump-
tion of un-informative priors, the main computation
then is the likelihood P(D/τ
i
). The procedure thus
searches for a set of plausible trees that are weighted
by their probabilities. In practice a specific tree
is modelled by M = {τ, υ,Q,γ}: topology, branch
lengths, DNA substitution parameters and gamma
shape parameter respectively.
2.1.2 Nucleotide Substitution Model
For a particular tree the transition probabilities from
root to all the leaf nodes needs to be defined. The
problem specification therefore includes a concrete
model of evolution. An evolutionary mechanism re-
sponsible for sequence change can be quantified in
terms of (rate of) nucleotide substitution as a sub-
stitution matrix Q. A number of models have been
proposed and it has been shown that different mod-
els are subsumed in the General Time Reversal (GTR)
model(Felsenstein, 2004). The GTR model states that
each character may be substituted for any other, and
this can be specified with a 4 × 4 instantaneous rate
matrix Q.
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
144
2.1.3 BayesPhylogenies
BayesPhylogenies implements a data driven MCMC
method for inferring phylogenetic trees incorporating
heterogeneous models of evolution in which P(D/τ
i
),
the likelihood, accounts for rate and pattern hetero-
geneity(Meade, 2011):
P(D | Q
γ
,τ) =
∏
i
∑
j
w
j
k
∑
k
P(D
i
| γ
k
Q
j
,τ) (2)
where j is the number of independent models of
evolution, w is a weighting vector of length j, which
sums to 1. γ is a discretised gamma distribution, with
k categories. The addition of rate and pattern hetero-
geneity requires j × k passes over the tree. A typi-
cal analysis can consists of 40 independent runs when
multiple data sets, chains and models are considered.
Current implementation require weeks of compu-
tational time to analyse a model run for 1k taxa on
1k core (Meade, 2011). Our goal is to advance solu-
tions to big data levels with emerging Exascale com-
putation — from analysis of 10k to 100k taxa and
beyond. We investigated scalability of BayesPhylo-
genies on Harvard’s Ozone, a 0.5 Petaflop, 15k cores
cluster as a ”Tier 3” pre-production configuration to
execute a Grand Challenge run that uses the full sys-
tem (RC-FAS, 2018b). Scalability analysis was per-
formed on big data sets of 3k and 5k fish taxa, con-
sisting of 11621 nucleotides at a number of levels:
• scalability analysis to replicate the U.K. runs for
Ozone.
• strong and weak scaling runs on larger nodes
(128) and an exploration of the optimal number
of threads per task.
• scaling characteristics for model complexity i.e,
patterns 4 vs 10.
• a full scale run on 12880 cores over 12 hours.
3 SCALABILITY ANALYSIS ON
HARVARD’S OZONE 12K
CORES
BayesPhylogenies incorporates a number of models
and applying a combination for a given data set will
magnify the computational complexity by orders of
magnitude. We investigated scalability for various
model configurations as discussed below. We use a
scalability run across Ozone and UK clusters as a ref-
erence point from which the relative weak scaling is
then evaluated.
BayesPhylogenies implements a hybrid strategy
with two-level SIMD(openMP) and SPMD(MPI) par-
allelisation for scalable performance on hierarchi-
cal parallel architectures. SPMD (MPI) partitions
the gene data (11621 nucleotides) into independent
blocks and a model parallelisation then computes the
two summation in equation 2 in parallel for each site
in the partition.
3.1 Scaling Comparison with U.K. Runs
The U.K. analysis is run on a 40 node configuration
with 12 cores per node (named SBS in figures). Since
convergence of MCMC can take months of compute
time for a 3k data set, scalability analysis is performed
by executing a fixed number of iterations, 20,000 and
with model parameters pattern=4 and gamma=4.
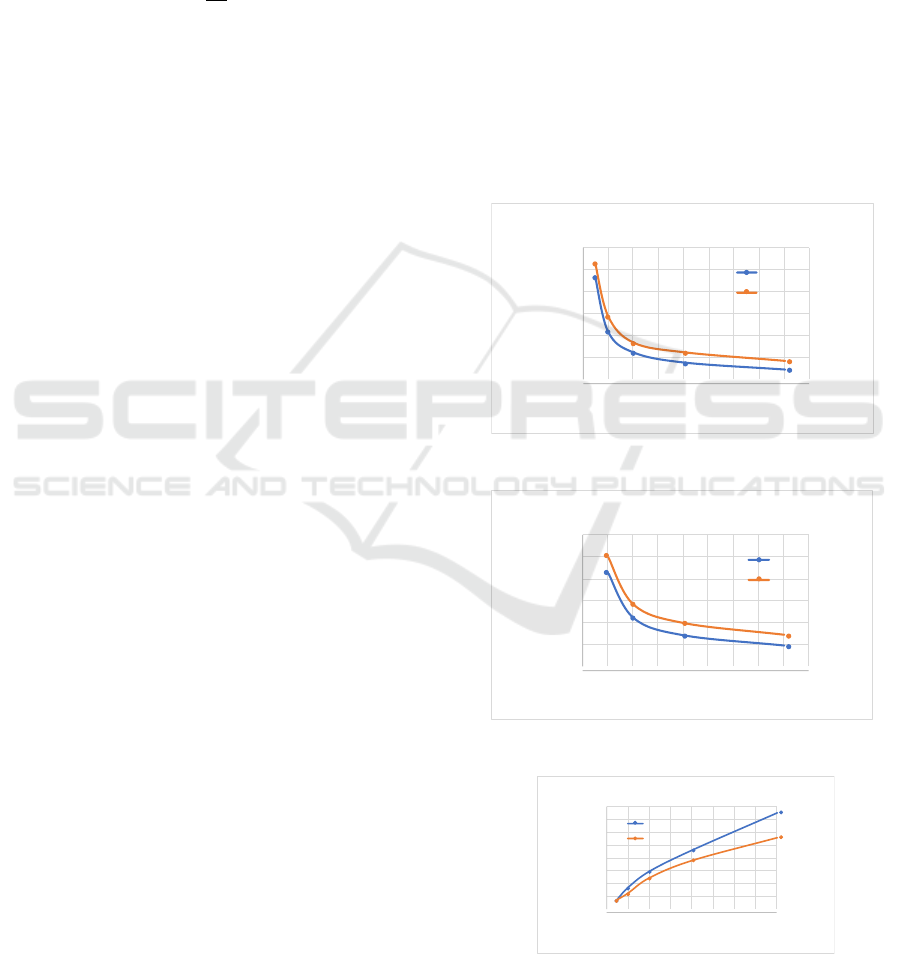
!"!!
#"!!
$!"!!
$#"!!
%!"!!
%#"!!
&!"!!
! ' ( $% $) %! %' %( &% &)
*+,-./0
123/ 0
&456+07538.8
9:2,/
;<;
Figure 1: a: Comparison of Ozone and SBS.
!"!!
#"!!
$!"!!
$#"!!
%!"!!
%#"!!
&!"!!
! ' ( $% $) %! %' %( &% &)
*+,-./0
123/ 0
#456+075.898
:;2,/
<=<
Figure 2: b: Comparison of Ozone and SBS.
!
"
#
$
%
&!
&"
&#
&$
! # % &" &$ "! "# "% '"
()**+,)
-.+*/
()**+,)0 .1023.1*
'45/)**+,)
645/)**+,)
Figure 3: Big data sets speedup.
On both architectures the software scales for 3k
and 5k data sets with similar speed-ups as shown in
Big Data Scalability of BayesPhylogenies on Harvard’s Ozone 12k Cores
145
figures 1, 2, 3. However Ozone has 32 cores per node
against 12 cores of SBS and hence similar MPI tasks
to OpenMP threads ratio are evaluated for a fair com-
parison. The best MPI, OpenMP combination is cho-
sen for comparison as MPI=2, OpenMP=6 for SBS
and 2 × Nodes MPI tasks on Ozone in the graphs
shown in figure 1. Although the speed-ups are mod-
est at the node level (16 for 32 nodes) as in figure 3,
the parallel efficiency drops significantly when mul-
tiple cores per node are taken into account. Hence,
simple metrics are not adequate to deduce scalability
limitations.
3.2 Strong and Weak Scaling on 4k
Cores
Here the interest is in a number of scaling characteris-
tics on Ozone with a large node (128) and core count
(4k) relative to SBS cluster. The runs are performed
for 40,000 iterations with default models parameters
(pattern=4, gamma=4) as above and a variant (pat-
tern=10) to evaluate model scaling.
1. Hybrid Parallelisation: In this we are interested
in tasks to threads ratios that gives the best perfor-
mance which can only be determined empirically
due to the data dependent nature of MCMC based
inference. The evaluation on 128 nodes gives an
optimal ratio of MPI tasks to OpenMP threads as
256:16 for 1k, 3k and 5k taxa. The hybrid scal-
ing however saturates at 256 MPI tasks due to in-
creased MPI communication overheads.
2. Strong Scaling: In strong scaling a parallel pro-
gram is defined as scaling linearly if the speed-up
equals the number of processing elements N. We
set the threads per task to the optimum value of
16 and varied the node count for a fixed workload
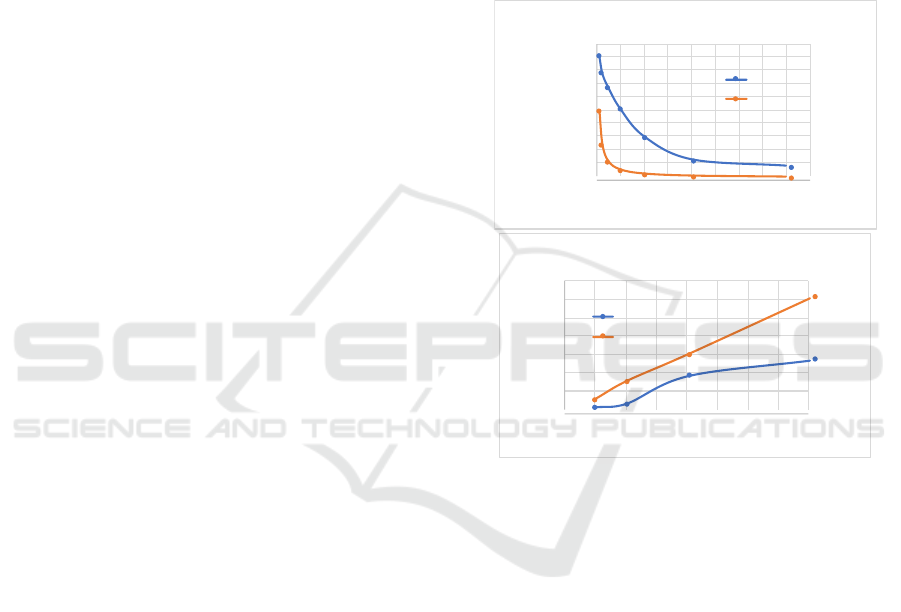
of 3k taxa. The strong scaling efficiency drops
below 50% beyond 20 nodes as shown in figure
4. This is largely due to limited parallelism of
model parallelisation (openMP) on a single node
(32 cores) combined with increase in communica-
tion overhead from a fine grain decomposition at
large node count (128).
3. Weak Scaling: Analysis was performed by dou-
bling the workload from 500, 1k, 3k to 5k taxa
whilst doubling the number of nodes 16, 32,
64 and 128. With 2 MPI tasks per node and
16 threads per task the execution times are as
in figure 4. The execution time increases due
to data dependent nature of bayesian phyloge-
nies analysis and the iterative nature of MCMC
sampling from large posterior distributions as the
taxa size increases (equation 2). Additionally, as
noted above the communication overheads begin
to dominate beyond 64 nodes.
4. Model Scaling: results of strong and weak scal-
ing analysis with increased model complexity by
setting pattern=10 (k in equation 2) is shown in
figure 4. Due to multiple passes in calculating
the likelihood the strong scaling effects are more
pronounced. Thus improving strong scaling for
multi-cores is an important parallel efficiency op-
timization for big data analysis.
!
"!
#!
$!
%!
&!
'!
(!
)!
*!
"!!
! "' $# %) '% )! *' ""# "#) "%%
+,-,../.0 /1123/4250,6070 810.34/,-0 62,.349
:8;/6
<=-849062,.3490
>,==/-4?%
>,==/-4?"!
!
"
#
$
%
&!
&"
&#
! &$ '" #% $# %! ($ &&" &"%
)*+,-./
012./3
4.5637859*+:3
;<#
;<&!
Figure 4: Strong and weak scaling for different models (p=4
and p=10).
3.3 Scaling at 12k Cores on Ozone
A full scale run to assess how well the parallelisa-
tion leverages the capacity of 12880 cores on 400
nodes was performed for the 3k fish data with de-
fault model parameters. With optimal threads of 16
per MPI task derived from above scalability analysis,
with 1600 MPI ranks the large scale run completed 7
million iterations in 12 hours. However in order to
achieve convergence of MCMC we need the analysis
to be run for 500M iterations for these types of big
data sets.
For parallel runs at this scale a number of factors
impact on the scaling. The parallel formulation be-
comes fine grained as each node now gets a smaller
fraction of the data set. This results in increased MPI
communication overheads and as in the previous anal-
ysis of strong scaling (figure 4) the efficiency drops
beyond 64 nodes. The iterative nature of MCMC sam-
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
146

pling, the schedules involved in perturbing the tree
topologies in computing likelihood and the non-linear
memory access patterns in propagating likelihood val-
ues on trees all contribute to lower efficiency. Scaling
to big data therefore requires radical approaches to
parallel algorithm design and optimizations, in par-
ticular in strong scaling techniques.
4 DISCUSSION
The evaluation study focuses on the scalability char-
acteristics of the Bayesian computational procedure
and its parallelisation and not the generation of final
trees. Hence the quality of output is not the focus of
the study as that requires the computation to be run
to convergence as noted earlier. However the study
provides insight into parallel algorithm implementa-
tion for any Bayesian phylogenetic analysis that is
MCMC based. Any parallelisation has to consider the
multi-level parallelisation available in heterogenous
massively parallel architectures and therefore has to
consider strong and weak scaling.
Two communication complexity, from data move-
ment across the memory hierarchy and across the dis-
tributed memory, impact on the scalability as shown.
Even though the current implementation already im-
plements multi-level SIMD/SPMD parallelisation the
data dependent nature of the computation requires
more complex program analysis and tranformation to
overcome scalability limitations.
To make a step change to the next generation of
exascale ready Computational Phylogenetics a num-
ber of models of parallelisation run-time optimiza-
tions and algorithmic adaptations will be required:
• A hierarchical SPMD parallelisation and radical
approaches to minimizing data movement in the
memory hierarchy are approaches to improving
strong scaling for tree-based computations(Kamil
and Yelick, 2014).
• Dynamic parallel computations with asyn-
chronous task parallelisation combined with
dynamic load balancing, MPI+PGAS or
MPI+OpenMPtask, with supporting run-time
for automatic thread migration across nodes are
alternative algorithm design formulations that can
enhance scalability(J. Hashmi, 2016).
• Radical scaling can also be obtained from fun-
damental algorithmic adaptations with better con-
vergence properties than MCMCMC.
The scalability study although uses fish data set
is generalisable from parallelisation and algorithmic
viewpoint with the emerging approaches discussed
above. Nevertheless, other factors such as evolution-
ary diameter of a particular data set should be con-
sidered in designing effective parallel algorithm if
it affects the convergence of MCMC computational
models. This aspect further re-enforces the require-
ment for data driven nature of applications to consider
both algorithmic and emerging strong and weak scal-
ing strategies in the implementation of computational
models for phylogenetics.
5 CONCLUSION
The performance of data dependent computational
models are characterized by an interplay of the al-
gorithm, architecture and data sets as shown by the
evaluation studies above. The scalability analysis pro-
vides insights into the importance of strong-scaling
for multi-core systems for parallel applications that
are memory bound. Unlike in data decomposition
of typical data parallel applications where workload
per processor can be characterised in terms of prob-
lem size, data dependent computations as in MCMC
bayesian inference pose considerable challenges in
optimizing the computations for high parallel effi-
ciency and in evaluating their performance with more
sophisticated metrics. The design of effective com-
putational procedures therefore needs to consider al-
gorithmic and problem domain characteristics as dis-
cussed above. Several alternative parallelisation and
algorithm strategies as proposed above are steps to-
wards realising next generation scalable models that
are ’exascale ready’. With sophisticated computa-
tional techniques this grand challenge problem can,
for the first time, be addressed at large scale.
ACKNOWLEDGEMENTS
Thanks to RC, FAS at Harvard for allocating Ozone
for this scalability study. Prof. Manjunathaiah was
a visiting faculty at IACS/SEAS, Harvard University,
USA, in 2017 during this research.
REFERENCES
Felsenstein, J. (2004). Inferring phylogenies. Sinauer As-
sociates, Inc.
J. Hashmi, K. Hamidouche, D. P. (2016). Enabling
performance efficient runtime support for hybrid
mpi+upc++ programming models.
Kamil, A. and Yelick, K. (2014). Hierarchical computation
in the spmd programming model. In Cas
,
caval, C. and
Big Data Scalability of BayesPhylogenies on Harvard’s Ozone 12k Cores
147

Montesinos, P., editors, Languages and Compilers for
Parallel Computing, pages 3–19, Cham. Springer In-
ternational Publishing.
Meade, A. (2011). Scalable methods for bayesian phyloge-
netic inference. In (unpublished). University of Read-
ing, U.K.
Pagel, M. and Meade, A. (2006). Bayesian analysis of cor-
related evolution of discrete characters by reversible-
jump markov chain monte carlo. The American Natu-
ralist, 167(6):808–825.
Pagel, M. and Meade, A. (2008). Modelling hetero-
tachy in phylogenetic inference by reversible-jump
markov chain monte carlo. Philosophical Transac-
tions of the Royal Society B: Biological Sciences,
363(1512):3955–3964.
RC-FAS ((retrieved 2018)a). Bayesphylogenies.
https://portal.rc.fas.harvard.edu/apps/modules/-
BayesPhylogenies.
RC-FAS ((retrieved 2018)b). Odyssey 3, the next gener-
ation. https://www.rc.fas.harvard.edu/odyssey-3-the-
next-generation/.
Salichos, L. and Rokas, A. (2013). Inferring ancient diver-
gences requires genes with strong phylogenetic sig-
nals. Nature, 497(7449):327.
Warnow, T. (2017). Computational challenges in construct-
ing the tree of life. In 2017 IEEE International Paral-
lel and Distributed Processing Symposium (IPDPS),
pages 1–1.
BIOINFORMATICS 2019 - 10th International Conference on Bioinformatics Models, Methods and Algorithms
148
