
Using a Fuzzy Decision Tree Ensemble for Tumor Classification from
Gene Expression Data
Jos
´
e M. Cadenas
1
, M. Carmen Garrido
1
, Raquel Mart
´
ınez
1
, David A. Pelta
2
and Piero P. Bonissone
3
1
Dept. of Information Engineering and Communications, Computer Faculty, University of Murcia, Murcia, Spain
2
Dept. of Computer Science and Artificial Intelligence, University of Granada, Granada, Spain
3
General Electric Global Research, One Research Circle, Niskayuna, New York, U.S.A.
Keywords:
Fuzzy Random Forest, Gene Selection, Gene Expression Data, Tumor Datasets.
Abstract:
Machine learning techniques are useful tools that can help us in the knowledge extraction from gene expression
data in biological systems. In this paper two machine learning techniques are applied to tumor datasets based
on gene expression data. Both techniques are based on a fuzzy decision tree ensemble and are used to carry
out the classification and selection of features on datasets. The classification accuracies obtained both when
we use all genes to classify and when we only use the selected genes are high. However, in this second case
the result also increases the interpretability of the solution provided by the technique. Additionally, the feature
selection technique provides a ranking of importance of genes and a partitioning of the domains of the genes.
1 TUMOR CLASSIFICATION
FROM GENE EXPRESSION
DATA
The challenge of cancer treatment has been to tar-
get specific therapies to pathogenetically distinct tu-
mor types, to maximize efficacy and minimize toxic-
ity. Improvements in cancer classification have thus
been central to advances in cancer treatment. Can-
cer classification is divided into two challenges: class
discovery and class prediction. Class discovery refers
to defining previously unrecognized tumor subtypes.
Class prediction refers to the assignment of particu-
lar tumor examples to already-defined classes. In the
early days, cancer classification has been relying on
subjective judgment from experienced pathologists.
When microarray technology was discovered began
to be applied to cancer diagnosis. The most important
application of the microarray technique is to discrimi-
nate the normal and cancerous tissue samples accord-
ing to their expression levels, identify a small subset
of genes that are responsible for the disease and to
discover potential drugs, (Ghoraia et al., 2012).
Experimental techniques based on oligonu-
cleotide or cDNA arrays now allow the expression
level of thousands of genes to be monitored in par-
allel (Alon et al., 1999). To use the full potential of
such experiments, it is important to develop the ability
to process and extract useful information from large
gene expression datasets.
Constantly improving gene expression profiling
technologies are expected to provide understanding
and insight into cancer related cellular processes.
Gene expression data is also expected to significantly
aid in the development of efficient cancer diagnosis
and classification platforms. Gene expression data
can help in better understanding of cancer. Normal
cells can evolve into malignant cancer cells through a
series of mutations in genes that control the cell cy-
cle, apoptosis, and genome integrity, to name only a
few. As determination of cancer type and stage is of-
ten crucial to the assignment of appropriate treatment
(Golub et al., 1999), a central goal of the analysis of
gene expression data is the identification of sets of
genes that can serve, via expression profiling assays,
as classification or diagnosis platforms.
Another important purpose of gene expression
studies is to improve understanding of cellular re-
sponses to drug treatment. Expression profiling as-
says performed before, during and after treatment, are
aimed at identifying drug responsive genes, indica-
tions of treatment outcomes, and at identifying poten-
tial drug targets (Clarke et al., 1999). More generally,
complete profiles can be considered as a potential ba-
sis for classification of treatment progression or other
trends in the evolution of the treated cells.
320
Cadenas J., Garrido M., Martínez R., A. Pelta D. and P. Bonissone P..
Using a Fuzzy Decision Tree Ensemble for Tumor Classification from Gene Expression Data.
DOI: 10.5220/0004658203200331
In Proceedings of the 5th International Joint Conference on Computational Intelligence (SCA-2013), pages 320-331
ISBN: 978-989-8565-77-8
Copyright
c
2013 SCITEPRESS (Science and Technology Publications, Lda.)

Data obtained from cancer related gene expression
studies typically consists of expression level mea-
surements of thousands of genes. This complexity
calls for data analysis methodologies that will effi-
ciently aid in extracting relevant biological informa-
tion. Previous gene expression analysis work em-
phasizes clustering techniques (nonsupervised classi-
fication), which aim at partitioning the set of genes
into subsets that are expressed similarly across differ-
ent conditions. On the other hand, supervised clas-
sification techniques (also called class prediction or
class discrimination) with the aim to assign examples
to predefined categories, (Golub et al., 1999; Diaz-
Uriarte and de Andr
´
es, 2006; Nitsch et al., 2010).
The objectives of supervised classification tech-
niques are: 1) to build accurate classifiers that enable
the reliable discrimination between different cancer
classes, 2) to identify biomarkers of diseases, i.e. a
small set of genes that leads to the correct discrimi-
nation between different cancer states. This second
purpose of supervised classification can be achieved
by classifiers that provide understandable results and
indicate which genes contribute to the discrimination.
Following this line, in this paper the goal is to
apply two techniques to classify and select features
to tumor datasets in order to carry out an analysis of
these datasets and to obtain the information that pro-
vide understandable results. We use the Fuzzy Ran-
dom Forest method (FRF) proposed in (Bonissone
et al., 2010; Cadenas et al., 2012a) and the Feature Se-
lection Fuzzy Random Forest method (FRF-fs) pro-
posed in (Cadenas et al., 2013).
This paper is organized as follows. First, in Sec-
tion 2 some techniques applied to gene expression
data reported in literature are briefly described. Next,
in Section 3, the applied methods are described. Then,
in Section 4 we perform an analysis of two tumor
datasets using these methods. Finally, in Section 5
remarks and conclusions are presented.
2 MACHINE LEARNING AND
GENE EXPRESSION DATA
In this section, we describe some of the machine
learning techniques used for the management of gene
expression data.
2.1 Cluster Analysis based Techniques
Clustering is one of the primary approaches to ana-
lyze such large amount of data to discover the groups
of co-expressed genes. In (Mukhopadhyaya and
Maulikb, 2009) an attempt to improve a fuzzy clus-
tering solution by using SVM classifier is presented.
In this regard, two fuzzy clustering algorithm, VGA
and IFCM have been used.
In (Alon et al., 1999) a clustering algorithm to or-
ganize the data in a binary tree is used. The algorithm
was applied to both the genes and the tissues, reveal-
ing broad coherent patterns that suggest a high degree
of organization underlying gene expression in these
tissues. Coregulated families of genes clustered to-
gether. Clustering also separated cancerous from non-
cancerous tissue.
In (Golub et al., 1999) a SOM to divide the
leukemia examples into cluster is used. First, they
applied a two-cluster SOM to automatically discov-
ering the two types of leukemia. Next, they applied
a four-cluster SOM. They subsequently obtained im-
munophenotype data on the examples and found that
the four classes largely corresponded to AML, T-
lineage ALL, B-lineage ALL, and B-lineage ALL, re-
spectively. The four-cluster SOM thus divided the ex-
amples along another key biological distinction.
In (Ben-Dor et al., 2000) a clustering based clas-
sifier is built. The clustering algorithm on which the
classifier is constructed is the CAST algorithm that
takes as input a threshold parameter t, which controls
the granularity of the resulting cluster structure, and
a similarity measure between the tissues. To classify
a example they cluster the training data and example,
maximizing compatibility to the labeling of the train-
ing data. Then they examine the labels of all elements
of the cluster the example belongs to and use a simple
majority rule to determine the unknown label.
2.2 Techniques for Feature Selection
and Supervised Classification
Discovering novel disease genes is still challenging
for constitutional genetic diseases (a disease involv-
ing the entire body or having a widespread array of
symptoms) for which no prior knowledge is available.
Performing genetic studies frequently result in large
lists of candidate genes of which only few can be fol-
lowed up for further investigation. Gene prioritiza-
tion establishes the ranking of candidate genes based
on their relevance with respect to a biological process
of interest, from which the most promising genes can
be selected for further analysis, (Nitsch et al., 2010).
This is a special case of feature selection, a well-
known problem in machine learning.
In (Golub et al., 1999) a procedure that uses
a fixed subset of “informative genes” is developed.
These “informative genes” are chosen based on their
correlation with the class distinction.
UsingaFuzzyDecisionTreeEnsembleforTumorClassificationfromGeneExpressionData
321

In (Diaz-Uriarte and de Andr
´
es, 2006), a Random
Forest ensemble is used to carry out the feature selec-
tion process for classification from gene expression
data. The technique calculates a measure of impor-
tance for each feature based on how the permutation
of the values of that feature in the dataset affects to
the classification of the out-of-bag (OOB) dataset of
each decision tree of ensemble (Breiman, 2001). Fol-
lowing this study, in (Genuer et al., 2010), a Random
forest ensemble which solves the problems existing in
(Diaz-Uriarte and de Andr
´
es, 2006) is proposed.
In (Duval and Hao, 2010) a study of classification
of gene expression data using metaheuristics is pre-
sented. The authors show that gene selection can be
casted as a combinatorial search problem, and conse-
quently be handled by these optimization techniques.
In (Nitsch et al., 2010), four different strategies to
prioritize candidate genes are proposed. These strate-
gies are based on network analysis of differential ex-
pression using distinct machine learning approaches
to determine whether a gene is surrounded by highly
differentially expressed genes in a functional associa-
tion or protein-protein interaction network.
Another work to select genes is proposed in
(Dagliyan et al., 2011). This paper shows that a sys-
tematic and efficient algorithm, mixed integer linear
programming based hyper-box enclosure (HBE) ap-
proach, can be applied to classification of different
cancer types efficiently.
3 CLASSIFICATION AND
FEATURE SELECTION BY
FUZZY RANDOM FOREST
In this section, we describe the methods that we will
use in this paper.
3.1 Fuzzy Random Forest for
Classification
We briefly describe the Fuzzy Random Forest (FRF)
ensemble proposed in (Bonissone et al., 2010; Cade-
nas et al., 2012a). FRF ensemble was originally pre-
sented in (Bonissone et al., 2010), and then extended
in (Cadenas et al., 2012a), to handle imprecise and
uncertain data. We describe the basic elements that
compose this FRF ensemble and the types of data that
are supported by this ensemble in both learning and
classification phases.
Fuzzy Random Forest Learning: Let E be a dataset.
FRF learning phase uses Algorithm 1 to generate the
FRF ensemble whose base classifier is a Fuzzy Deci-
sion Tree (FDT). Algorithm 2 shows the FDT learning
algorithm, (Cadenas et al., 2012b).
Algorithm 1: FRFlearning.
1: Input: E, Fuzzy Partition; Output: FRF
2: begin
3: repeat
4: Take a random sample of |E| examples with replace-
ment from the dataset E
5: Apply Algorithm 2 to the subset of examples ob-
tained in the previous step to construct a FDT
6: until all FDTs are built to constitute the FRF ensemble
7: end
Algorithm 2: FDecisionTree.
1: Input: E, Fuzzy Partition; Output: FDT
2: begin
3: Start with the examples in E with values
χ
Fuzzy Tree,root
(e) = 1 to all examples with a sin-
gle class and replicate the examples with set-valued
class and initialize their weight according to the
available knowledge about their class
4: Let A be the feature set (all numerical features are par-
titioned according to the Fuzzy Partition)
5: repeat
6: Choose a feature to the split at the node N
7: loop
8: Make a random selection of features from the set A
9: Compute the information gain for each selected
feature using the values χ
Fuzzy Tree,N
(e) of each e
in node N taking into account the function µ
simil(e)
for the cases required
10: Choose the feature such that information gain is
maximal
11: end loop
12: Divide N in children nodes according to possible se-
lected feature outputs in the previous step and re-
move it from the set A. Let E
n
be the dataset of each
child node
13: until the stopping criteria is satisfied
14: end
Algorithm 2 has been designed so that the FDTs
can be constructed without considering all the fea-
tures to split the nodes. Algorithm 2 is an algorithm
to construct FDTs where the numerical features have
been discretized by a fuzzy partition. The domain of
each numerical feature is represented by trapezoidal
fuzzy sets, F
1
, . . . , F
f
so each internal node of the
FDTs, whose division is based on a numerical feature,
generates a child node for each fuzzy set of the parti-
tion. Moreover, Algorithm 2 uses a function, denoted
by χ
t,N
(e), that indicates the degree with which the
example e satisfies the conditions that lead to node N
of FDT t. Each example e is composed of features
which can be crisp, missing, interval, fuzzy values
IJCCI2013-InternationalJointConferenceonComputationalIntelligence
322
belonging (or not) to the fuzzy partition of the fea-
ture. Furthermore, we allow the class feature to be
set-valued. These examples (according to the value
of their features) have the following treatment:
• Each example e used in the training of the FDT t
has assigned an initial value χ
t,root
(e). If an ex-
ample has a single class this value is 1. If an
example has a set-valued class, it is replicated
with a weight according to the available knowl-
edge about the classes.
• According to the membership degree of the exam-
ple e to different fuzzy sets of partition of a split
based on a numerical feature:
– If the value of e is crisp, the example e
may belong to one or two children nodes,
i.e., µ
f uzzy set partition
(e) > 0. In this case
χ
t,childnode
(e) = χ
t,node
(e) · µ
f uzzy set partition
(e).
– If the value of e is a fuzzy value matching with
one of the sets of the fuzzy partition of the fea-
ture, e will descend to the child node associ-
ated. In this case, χ
t,childnode
(e) = χ
t,node
(e).
– If the value of e is a fuzzy value different from
the sets of the fuzzy partition of the feature, or
the value of e is an interval value, we use a sim-
ilarity measure, µ
simil
(·), that, given the feature
“Attr” to be used to split a node, measures the
similarity between the values of the fuzzy par-
tition of the feature and fuzzy values or inter-
vals of the example in that feature. In this case,
χ
t
,
childnode
(e) = χ
t
,
node
· µ
simil
(e).
– When the example e has a missing value, the
example descends to each child node node
h
,
h = 1, . . . , H
i
with a modified value proportion-
ately to the weight of each child node. The
modified value for each node
h
is calculate as
χ
node
h
(e) = χ
node
(e) ·
T χ
node
h
T χ
node
where T χ
node
is
the sum of the weights of the examples with
known value in the feature i at node node and
T χ
node
h
is the sum of the weights of the exam-
ples with known value in the feature i that de-
scend to the child node node
h
.
Fuzzy Random Forest Classification
The fuzzy classifier module operates on FDTs of the
FRF ensemble using one of these two possible strate-
gies: Strategy 1 - Combining the information from the
different leaves reached in each FDT to obtain the de-
cision of each individual FDT and then applying the
same or another combination method to generate the
global decision of the FRF ensemble; and Strategy 2
- Combining the information from all leaves reached
from all FDTs to generate the global decision of the
3.2 Fuzzy Random Forest for Feature
Selection
The FRF-fs method (Cadenas et al., 2013) is classi-
fied as a hybrid method that combines the filter and
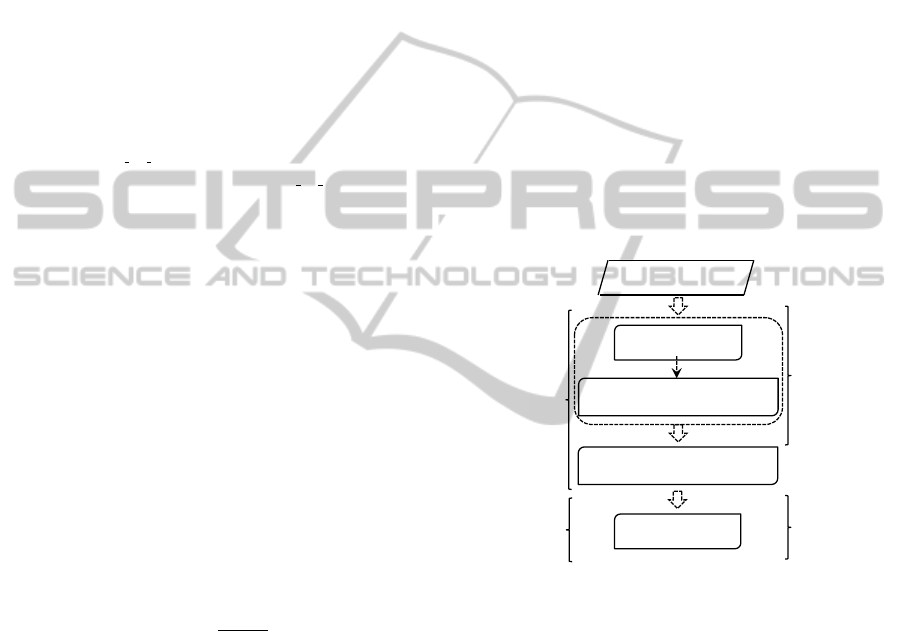
wrapper methods. The framework (Fig. 1) consists
of main steps: (1) Scaling and discretization process
of the feature set; and feature pre-selection using the
discretization process; (2) The feature pre-selection
ranking process using information given by Fuzzy
Random Forest ensemble; and (3) Wrapper feature se-
lection using a classification technique. Starting from
the ordered features, this wrapper method constructs
an ascending sequence of sets of candidate features,
by invoking and testing the features stepwise. The
different feature subsets obtained by this process are
evaluated by a machine learning method. In each step,
the method obtains information useful to the user:
pre-selected feature subset, feature subsets ranking
and optimal feature subset.
Dataset Feature set
Ranking process of features
Feature subsets
Data preprocess
Obtaining subset of features
Preselection and Ranking
Optimal feature
subset
WRAPPER METHOD
FILTER METHOD
Figure 1:: Framework of FRF-fs.
In the filter method, we use the method proposed
in (Cadenas et al., 2012b). From the feature subset
and the dataset obtained with the filter method, we
apply FRF method. Once FRF ensemble has been ob-
tained, we have all the information about each FDT.
Algorithm 3 describes how information provided for
each FDT of the ensemble is compiled and used to
measure the importance of each feature.
More specifically, the information we get from
each FDT t for each feature a is the following:
• Information gain of node N for the feature a
(IG
Na
) where the feature a has been selected as
the best candidate to split it.
• Depth level of node N (P
Na
) where feature a has
been selected as the best candidate to split it.
• Classification accuracy Acc
t
of FDT t when clas-
sifying the dataset OOB
t
.
UsingaFuzzyDecisionTreeEnsembleforTumorClassificationfromGeneExpressionData
323

Algorithm 3: INFFRF Information of the FRF.
1: Input: E, Fuzzy Partition, T N; Output: INF
2: begin
3: Building a Fuzzy Random Forest (Algorithm 1 - 3.1)
4: for each FDT t=1 to T N of the FRF ensemble do
5: Save the feature a chosen to split each node N, in-
formation gain of node, IG
Na
, and the depth of that
node P
Na
, in INF
a
.
6: Obtain the classification accuracy Acc
t
of the FDT t
with its corresponding OOB
t
dataset.
7: end for
8: end
Algorithm 4 details how the information INF ob-
tained from the FRF ensemble is combined to obtain
an importance measure of the features where p
i
is the
weight we assign to feature a depending on the place
where it appears in the FDT t. After the information
is combined, the output of this algorithm is a matrix
(IMP) where for each FDT t and for each feature a,
the importance value obtained in the FDT t for the
feature a is stored.
Algorithm 4: IMPFRF Combining information INF.
1: Input: INF, T N; Output: IMP
2: begin
3: for each FDT t=1 to T N do
4: for each feature a=1 to |Attr| do
5: for all nodes N where feature a appears do
6: if P
Na
= i then
7: IMP
ta
= IMP
ta
+ p
i
· IG
Na
with i ≥ 0 and
P
rootnode
= 0
8: end if
9: end for
10: for each feature a=1 to |Attr| do
11: IMP
ta
=
(
IMP
ta
−min(IMP
t
)
max(IMP
t
)−min(IMP
t
)
)
· OOB
t
12: end for
13: The vector IMP
t
is ordered in descending or-
der, IMP
t
σ
t
, where σ
t
is the permutation obtained
when ordering IMP
t
14: end for
15: end for
16: end
The idea behind the measure of importance of
each feature is that it uses the features of the FDTs
obtained and the decision nodes built with them in
the following way. The importance of a feature is de-
termined by its depth in a FDT. Therefore a feature
that appears on the top of a FDT is more important
in that FDT than another feature that appears in the
lower nodes. And, a FDT that has a classification ac-
curacy greater than another to classify the correspond-
ing OOB (dataset independent of the training dataset)
is a better FDT. The final decision is agreed by the
information obtained for all FDTs.
As a result of Algorithm 4, we obtain for each
FDT of FRF ensemble an importance ranking of fea-
tures. Specifically, we will have T N importance rank-
ings for each feature a. Applying an operator OWA,
we add them into one ranking. This final ranking in-
dicates the definitive importance of the features.
OWA operators (Ordered Weighted Averaging)
were introduced by Yager in 1988, (Yager, 1988).
OWA operators are known as compensation opera-
tors. They are aggregation operators of numerical in-
formation that consider the order of the assessments
that will be added. In our case, we have T N ordered
sets. Given a weight vector W , the vector RANK rep-
resents the ranking of the pre-selected feature subset
and is obtained as follows (the vector RANK is or-
dered in descending order: RANK
σ
):
OWAIMP
t
= W ·IMP
t
σ
t
, for t = 1, . . . , T N
RANK
a
=
T N
∑
t=1
OWAIMP
tσ
t
(a)
, for a = 1, . . . , |A|
3.3 Wrapper for Feature Final Selection
Once the ranking of the pre-selected feature subset,
RANK
σ
, is obtained, we have to find an optimal sub-
set of features. One option to search the optimal sub-
set is by adding a single feature at a time following a
process that uses RANK
σ
. The several feature subsets
obtained by this process are evaluated by a machine
learning method that supports low quality data (called
Classi f ier
LQD
) with a process of cross-validation.
The detailed process of the proposed wrapper method
is shown in Algorithm 5.
Starting from the ordered feature pre-selected,
construct an ascending sequence of FRF models, by
invoking and testing the features stepwise. We per-
form a sequential feature introduction in two phases:
• In the first phase two feature subsets are built: the
feature subsets CF
base
and CF
comp
. A feature f
i
is
added to the CF
base
subset only if the decrease of
the error rate using the features of CF
base
∪ { f
i
}
subset exceeds a threshold δ
1
. The idea is that the
error decrease by adding f
i
must be significant for
that feature to belong to the CF
base
subset. If when
we classify using the subset CF
base
∪{ f
i
}, an error
decrease smaller than a threshold δ
1
or an error
increase smaller than a threshold δ
2
is obtained,
f
i
becomes part of the subset CF
comp
.
• The second phase starts with both CF
base
and
CF
comp
sets. We fix CF
base
and add feature sub-
groups from CF
comp
to build several FRF mod-
els. This phase determines the final feature set
with minimum error according to the conditions
IJCCI2013-InternationalJointConferenceonComputationalIntelligence
324

reflected on line 22 of Algorithm 5. These con-
ditions are interpreted as “select the subset that
decrements the error in an amount over threshold
δ
3
or decrements the error in an amount below δ
3
but using a smaller number of features.”
Algorithm 5: Wrapper method.
1: Input: E, candidate feature set C F and information
system RANK
σ
; Output: C F
opt
selected feature set
2: begin
3: CF
comp
= {} and CF
base
= { f
1
} where f
1
is the first
feature of RANK
σ
4: ERR
1
= Classi f ier(E,CF
base
) using cross-validation,
BE = ERR
1
5: for each f
i
∈ CF, with i = 2, . . . , |CF| in the order de-
termined by RANK
σ
do
6: ERR
B
= Classi f ier
LQD
(E,CF
base
∪ { f
i
}) using
cross-validation
7: if (BE − ERR
B
) > δ
1
then
8: CF
base
= CF
base
∪ { f
i
}
9: else
10: if (ERR
B
− BE) < δ
2
then
11: CF
comp
= CF
comp
∪ { f
i
}
12: end if
13: end if
14: end for
15: CF
aux
= CF
base
16: for each f
i
∈ CF
comp
, with i = 1, . . . , |CF
comp
| in the
order determined by RANK
σ
do
17: B = CF
base
, ST OP = 0, j = i
18: while (STOP < δ
2
) and ( j ≤ |CF
comp
|) do
19: B = B ∪ { f
j
}
20: ERR
B
= Classi f ier
LQD
(D, B) using cross-
validation
21: if ((BE −ERR
B
) ≥ δ
3
) or (0 ≤ (BE −ERR
B
) < δ
3
and |CF
aux
| > |B|) then
22: CF
aux
= B, BE = ERR
B
23: else
24: if (ERR
B
− BE) > δ
2
then
25: ST OP = (ERR
B
− BE)
26: end if
27: end if
28: j = j + 1
29: end while
30: end for
31: Return: CF
opt
= CF
aux
32: end
4 FRF AND TUMOR
CLASSIFICATION
In this section we examine the performance of the
FRF ensemble for classification and feature selection
from gene expression data.
4.1 Gene Expression Data
In this section, we describe the two datasets that we
will analyze. The first dataset involves comparing tu-
mor and normal examples of the same tissue, while
the second dataset involves examples from two vari-
ants of the same disease.
4.1.1 Colon Cancer and Leukemia Datasets
Colon tumor is a disease in which cancerous growths
are found in the tissues of the colon epithelial cells.
The Colon dataset contains 62 examples. Among
them, 40 tumor biopsies are from tumors (labeled
as “negative”) and 22 normal (labeled as “positive”)
biopsies are from healthy parts of the colons of the
same patients. The final assignments of the status of
biopsy examples were made by pathological exami-
nation. The total number of genes to be tested is 2000
(Alon et al., 1999).
In the 1960s was provided the first basis for clas-
sification of acute leukemias into those arising from
lymphoid precursors (acute lymphoblastic leukemia,
ALL) or from myeloid precursors (acute myeloid
leukemia, AML). The Leukemia dataset is a collec-
tion of expression measurements reported by (Golub
et al., 1999). The dataset contains 72 examples. These
examples are divided to two variants of leukemia:
25 examples of acute myeloid leukemia (AML) and
47 examples of acute lymphoblastic leukemia (ALL).
The source of the gene expression measurements was
taken from 63 bone marrow examples and 9 pe-
ripheral blood examples. Gene expression levels in
these 72 examples were measured using high density
oligonucleotide microarrays. The expression levels of
7129 genes are reported.
4.2 Estimating Prediction Errors
We apply the cross-validation method to evaluate the
prediction accuracy of the classification method. To
apply this method, we partition the dataset E into k
sets of examples, C
1
, . . . ,C
k
. Then, we construct a
data set D
i
= E −C
i
, and test the accuracy of a model
obtained from D
i
on the examples in C
i
. We estimate
the accuracy of the method by averaging the accuracy
over the k cross-validation trials.
There are several possible choices of k. A com-
mon approach is to set k =number of examples. This
method is known as leave one out cross validation
(LOOCV). We will use the LOOCV method.
Although our purpose is not to compare the results
with other methods, as a sample, in Tables
1
1 and 2 we
1
The results marked with A, B and C are obtained from
UsingaFuzzyDecisionTreeEnsembleforTumorClassificationfromGeneExpressionData
325
show the accuracy estimates for the different meth-
ods applied to the two datasets. The results obtained
in (Diaz-Uriarte and de Andr
´
es, 2006; Genuer et al.,
2010) are calculated with the .632+bootstrap method,
and the Leukemia dataset has 38 examples and 3051
features.
Table 1: Accuracy of different methods on Colon dataset.
Correct Unclassified
Clustering
A
88.70 0.00
Nearest Neighbor
A
80.60 0.00
SVM, linear kernel
A
77.40 9.70
SVM, quad. kernel
A
74.20 11.30
Boosting, 100 iter.
A
72.60 9.70
NN.vs
B
84.20 0.00
RF.du (s.e.=0)
B
84.10 0.00
RF.ge
C
91.70 0.00
FRF 91.94 0.00
Table 2: Acc. of different methods on Leukemia dataset.
Correct Unclassified
Nearest Neighbor
A
91.60 0.00
SVM, linear kernel
A
93.00 5.60
SVM, quad. kernel
A
94.40 4.20
Boosting, 100 iter.
A
95.80 1.40
NN.vs
B
44.40 0.00
RF.du (s.e.=1)
B
92.30 0.00
RF.ge
C
99.00 0.00
FRF 98.61 0.00
Estimates of classification accuracy give only a
partial insight on the performance of a method. Also,
we treat all errors as having equal penalty. In the
problems we handle, however, errors have asymmet-
ric weights. We distinguish false positive error - nor-
mal tissues classified as tumor, and false negative er-
rors - tumor tissues classified as normal. In diagnostic
applications, false negative errors can be detrimental,
while false positives may be tolerated.
ROC curves are used to evaluate the “power”
of a classification method for different asymmetric
weights (Brandley, 1997; Hanley and McNeil, 1982).
Since the area under the ROC curve (denoted by
AUC) is a portion of the area of the unit square, its
value will always be between 0.0 and 1.0. A real-
istic classifier should not have an AUC less than 0.5
(area under the diagonal line between (0,0) and (1,1)).
The AUC has an important statistical property: the
AUC of a classifier is equivalent to the probability
that the classifier will rank a randomly chosen posi-
tive instance higher than a randomly chosen negative
(Ben-Dor et al., 2000; Diaz-Uriarte and de Andr
´
es, 2006;
Genuer et al., 2010), respectively
instance. This is equivalent to the Wilcoxon test of
ranks (Hanley and McNeil, 1982).
The confusion matrixes obtained by applying FRF
to the two datasets are shown in Table 3.
Table 3: Confusion Matrixes obtained with FRF.
Colon Leukemia
actual value actual value
1 0 ALL AML
prediction 1 37 2 ALL 46 0
outcome 0 3 20 AML 1 25
Confusion matrix of Colon dataset shows five er-
rors, and a Specificity of 0.9091 and Sensibility of
0.9250. Confusion matrix of Leukemia dataset shows
one error, and a Specificity of 1.0 and Sensibility of
0.9787.
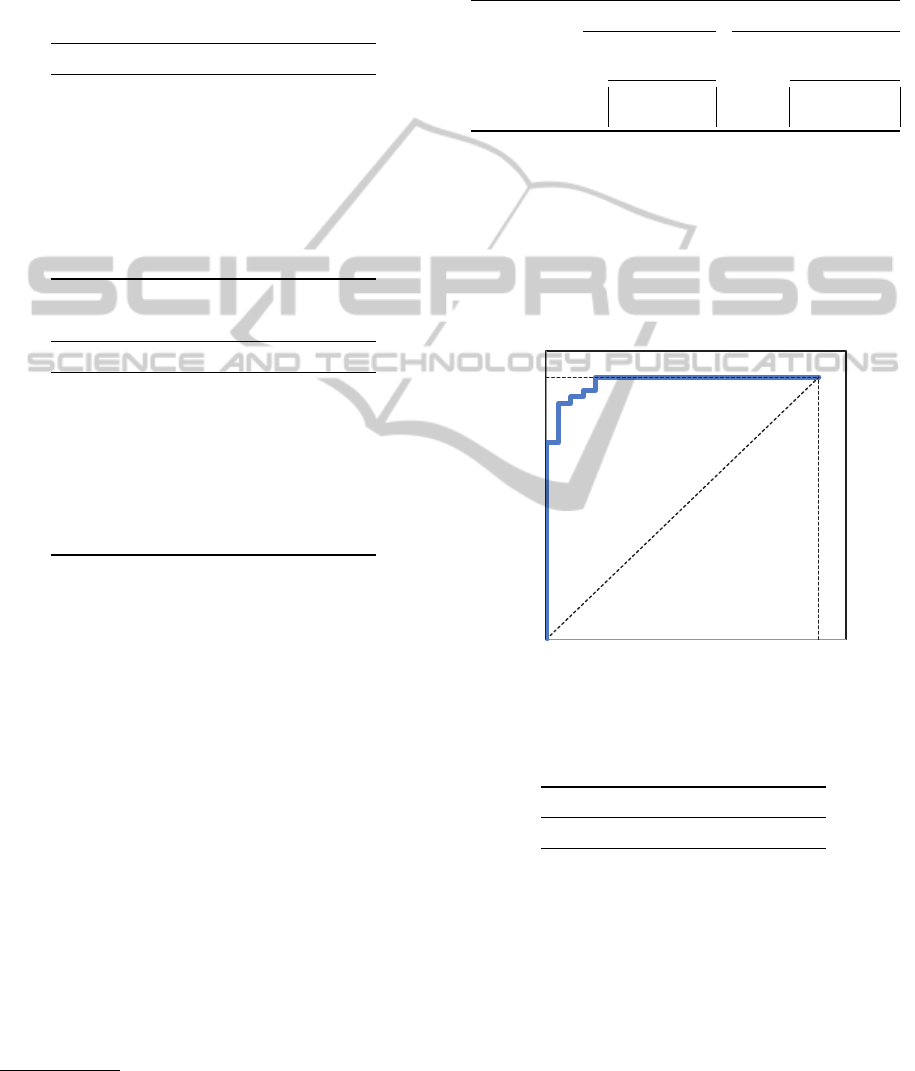
ROC curves are shown in Figures 2 and 3 and
AUC values in Table 4.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 1.1
sensitivity
1-specificity
Figure 2: Colon: ROC curve with all features.
Table 4: AUC values for Colon and Leukemia datasets.
Colon Leukemia
AUC values 0.9761 0.9991
4.3 Gene Selection
It is clear that the expression levels of many of the
genes in our datasets are irrelevant to the distinction
between tumors. Taking such genes into account dur-
ing classification increases the dimensionality of the
classification problem, presents computational diffi-
culties, and introduces noise to the process. Another
issue with a large number of genes is the interpretabil-
ity of the results. If our methods to distinguish tumor
IJCCI2013-InternationalJointConferenceonComputationalIntelligence
326

0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 1.1
sensitivity
1-specificity
Figure 3: Leukemia: ROC curve with all features.
from normal tissues is encoded in the expression lev-
els of few genes, then we might be able to understand
the biological significance of these genes.
Thus, it is crucial to recognize whether a small
number of genes can suffice for good classification.
The gene expression datasets are problematic in that
they contain a large number of genes (features) and
thus methods that search over subsets of features can
be expensive. Moreover, these datasets contain only a
small number of examples, so the detection of irrele-
vant genes can suffer from statistical instabilities.
4.3.1 Significance of a Gene and Ranking
The FRF-fs method (Cadenas et al., 2013) to feature
selection obtains a feature ranking based on an im-
portance measurement of each feature, and from that
ranking, an optimal feature subset. The vector RANK
(see Subsection 3.2) contains the importance measure
of the features. In Tables 9 and 10 (in Appendix 5)
a portion of that ranking of features and their impor-
tance values is shown.
4.3.2 Gene Prioritization in Cancer Data
In the final phase of the FRF-fs method (Cadenas
et al., 2013) an optimal feature subset is obtained.
In the Colon dataset the optimal feature subset is
{419, 765, 824, 1168, 513, 1772, 571, 1546, 1423,
1761, 1939, 1990, 377, 1668, 1346, 1586, 548, 474,
802, 1867}. In addition, to give more interpretability,
FRF-fs method obtains a feature partition. In Table
11 (in Appendix 5) we show the partition obtained for
this optimal features subset. The first column shows
the gene number while the second one shows the dif-
ferent partitions for this gene.
In the Leukemia dataset the optimal feature subset
is {3252, 4847, 2288, 2354, 6041, 6376, 4644}. In
Table 12 (in Appendix 5) we show the partition ob-
tained for this optimal features subset.
In Tables 13 and 14 (in Appendix 5) we show a
description of the selected genes (features) by FRF-fs
method. The first column shows the importance value
of each gene, the second one the gene number and the
third the description of it.
4.3.3 Classifying with Selected Subsets
Now, the classification procedure is applied using the
training data restricted to the subset of selected genes.
In Tables 5 and 6 we show the accuracy estimates
for the different methods applied to the two datasets
with the selected features.
Table 5: Accuracy with/without selected features for Colon
dataset.
FRF
All features Sel. features
Correct Unclassified Correct Unclassified
91.40 0.00 93.55 0.00
Table 6: Accuracy with/without selected features for
Leukemia dataset.
FRF
All features Sel. features
Correct Unclassified Correct Unclassified
98.61 0.00 98.61 0.00
The confusion matrixes obtained by applying FRF to
the two datasets with the selected features are shown
in Table 7.
Table 7: Confusion Matrixes obtained with FRF using se-
lected features.
Colon Leukemia
actual value actual value
1 0 ALL AML
prediction 1 38 2 ALL 46 1
outcome 0 2 20 AML 0 25
Confusion matrix of Colon dataset shows four er-
rors, and a Specificity of 0.9091 and Sensibility of
0.9500. Confusion matrix of Leukemia dataset shows
one error, and a Specificity of 0.9600 and Sensibility
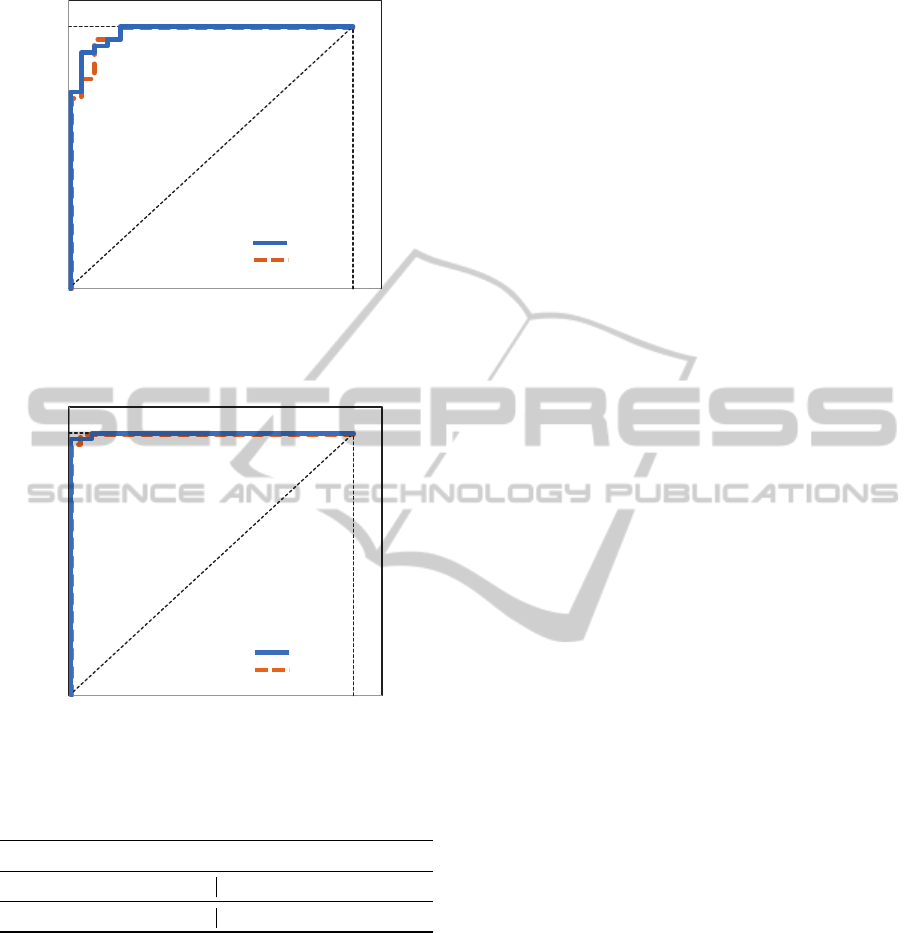
of 1.0. ROC curves are shown in Figures 4 and 5.
AUC values (Table 8) are compared with the obtained
when using all features.
Following the methods proposed in (Hanley and
McNeil, 1982; DeLong et al., 1988), we conclude that
UsingaFuzzyDecisionTreeEnsembleforTumorClassificationfromGeneExpressionData
327
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 1.1
sensitivity
1-specificity
all
selec.
Figure 4: Colon: ROC curve with all/selected features.
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
1.1
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1 1.1
sensitivity
1-specificity
all
selec.
Figure 5: Leukemia: ROC curve with all/selected features.
Table 8: AUC values.
Colon Leukemia
all features sel. features all features sel. features
0.9761 0.9710 0.9991 0.9987
there are no significant differences between the results
obtained when using all features or the selected ones.
We can therefore conclude that the selection of
features does not cause loss of accuracy but signifi-
cantly decreases the number of features.
5 CONCLUSIONS
In this paper we have applied a fuzzy decision tree en-
semble to tumor datasets with gene expression data.
On the one hand, we have applied the ensemble to
the classification of examples described by the set of
all features. On the other hand, we have applied the
ensemble to select a gene subset and to classify exam-
ples only described with the selected genes. The clas-
sification accuracies, in both cases, are high. These
results are validated statistically by the ROC curve
and AUC area.
When we work with a fuzzy decision tree ensem-
ble, in addition to achieve good results, these one are
provided in a highly interpretable way.
As part of the solution, the method provides a par-
tition of numerical features of the problem and a rank-
ing of importance of these features which permits the
identification of sets of genes that can serve as classi-
fication or diagnosis platforms.
ACKNOWLEDGEMENTS
Supported by the projects TIN2011-27696-C02-01
and TIN2011-27696-C02-02 of the Ministry of Econ-
omy and Competitiveness of Spain. Thanks also
to “Agencia de Ciencia y Tecnolog
´
ıa de la Regi
´
on
de Murcia” (Spain) for the support given to Raquel
Mart
´
ınez by the scholarship program FPI.
REFERENCES
Alon, U., Barkai, N., Notterman, D. A., Gish, K., Ybarra,
S., Mack, D., and Levine, A. J. (1999). Broad patterns
of gene expression revealed by clustering analysis of
tumor and normal colon tissues probed by oligonu-
cleotide arrays. Proc Natl Acad Sci U S A., 96:6745–
6750.
Ben-Dor, A., Bruhn, L., Friedman, N., Nachman, I.,
Schummer, M., and Yakhini, Z. (2000). Tissue clas-
sification with gene expression profiles. Journal of
Computational Biology, 7:559–583.
Bonissone, P. P., Cadenas, J. M., Garrido, M. C., and
D
´
ıaz-Valladares, R. A. (2010). A fuzzy random for-
est. International Journal of Approximate Reasoning,
51(7):729–747.
Brandley, A. P. (1997). The use of the area under the
roc curve in the evaluation of machine learning algo-
rithms. Pattern Recognition, 30(7):1145–1159.
Breiman, L. (2001). Random forests. Machine Learning,
43:5–32.
Cadenas, J. M., Garrido, M. C., and Mart
´
ınez, R. (2013).
Feature subset selection filter-wrapper based on low
quality data. Expert Systems with Applications, 40:1–
10. doi:10.1016/j.eswa.2013.05.051.
Cadenas, J. M., Garrido, M. C., Mart
´
ınez, R., and Bonis-
sone, P. P. (2012a). Extending information processing
IJCCI2013-InternationalJointConferenceonComputationalIntelligence
328

in a fuzzy random forest ensemble. Soft Computing,
16(5):845–861.
Cadenas, J. M., Garrido, M. C., Mart
´
ınez, R., and Bonis-
sone, P. P. (2012b). Ofp class: a hybrid method to
generate optimized fuzzy partitions for classification.
Soft Computing, 16:667–682.
Clarke, P., George, M., Cunningham, D., Swift, I., and
Workman, P. (1999). Analysis of tumor gene expres-
sion following chemotherapeutic treatment of patients
with bowel cancer. In Proc. Nature Genetics Microar-
ray Meeting, pages 39–39, Scottsdale, Arizona.
Dagliyan, O., Uney-Yuksektepe, F., Kavakli, I. H., and
Turkay, M. (2011). Optimization based tumor classi-
fication from microarray gene expression data. PLoS
One, 6:e14579.
DeLong, E. R., DeLong, D. M., and Clarke-Pearson, D. L.
(1988). Comparing the areas under two or more corre-
lated receiver operating characteristic curves: A non-
parametric approach. Biometrics, 44:837–845.
Diaz-Uriarte, R. and de Andr
´
es, S. A. (2006). Gene selec-
tion and classification of microarray data using ran-
dom forest. BMC Bioinformatics, 7(3).
Duval, B. and Hao, J.-K. (2010). Advances in metaheuris-
tics for gene selection and classification of microarray
data. Briefings in Bioinformatics, 11(1):127–141.
Genuer, R., Poggi, J.-M., and Tuleau-Malot, C. (2010).
Variable selecting using random forest. Pattern
Recognition Letters, 31(14):2225–2236.
Ghoraia, S., Mukherjeeb, A., and Duttab, P. K. (2012).
Gene expression data classification by vvrkfa. Pro-
cedia Technology, 4:330–335.
Golub, T. R., Slonim, D. K., Tamayo, P., Huard, C., Gaasen-
beek, M., Mesirov, J. P., Coller, H., Loh, M., Down-
ing, J., Caligiuri, M., Bloomfield, C., and Lander, E.
(1999). Molecular classification of cancer: Class dis-
covery and class prediction by gene expression moni-
toring. Science, 286:531–537.
Hanley, J. A. and McNeil, B. J. (1982). The meaning and
use of the area under a receiver operating characteris-
tic (roc) curve. Radiology, 143:29–36.
Mukhopadhyaya, A. and Maulikb, U. (2009). Towards
improving fuzzy clustering using support vector ma-
chine: Application to gene expression data. Pattern
Recognition Pattern Recognition, 42:2744–2763.
Nitsch, D., Gonzalves, J. P., Ojeda, F., de Moor, B., and
Moreau, Y. (2010). Candidate gene prioritization
by network analysis of differential expression using
machine learning approaches. BMC Bioinformatics,
11:460.
Yager, R. R. (1988). On ordered weighted averaging ag-
gregation operators in multicriteria decision making.
IEEE transactions on Systems, Man and Cybernetics,
18:183–190.
APPENDIX
Ranking and Partitions of Datasets
Table 9: Features Ranking in Colon dataset.
Ranking Fe. n.
1 35.6266 419
2 17.0359 765
3 15.6419 1635
4 13.5216 824
5 13.4986 1168
6 13.4898 513
7 9.6363 1772
8 7.2361 571
9 7.0409 1546
10 6.8134 1423
11 6.7085 1761
12 6.6085 1939
13 6.4989 1990
14 5.9908 377
15 4.6654 1668
16 4.0917 1346
17 3.1929 1586
18 2.3743 548
19 2.0175 474
20 1.8373 802
21 1.7315 1867
.. ..... ...
Table 10: Features Ranking in Leukemia dataset.
Ranking Fe. n.
1 31.2849 3252
2 30.1804 1882
3 30.1763 1834
4 26.5833 4847
5 23.9430 2288
6 13.5707 2354
7 13.1465 6041
8 9.8707 6376
9 4.8665 4644
10 1.4004 3623
.. ..... ...
UsingaFuzzyDecisionTreeEnsembleforTumorClassificationfromGeneExpressionData
329

Table 11: Features Partition in Colon dataset.
Fe.n. Partitions
377 (0,0,0.4046,0.5246) (0.4046,0.5246,1,1)
419 (0,0,0.6981,0.7140) (0.6981,0.7140,0.7241,0.7256) (0.7241,0.7256,1,1)
474 (0,0,0.8360,0.9194) (0.8360,0.9194,1,1)
513 (0,0,0.5625,0.5657) (0.5625,0.5657,1,1)
548 (0,0,0.7852,0.9132) (0.7852,0.9132,1,1)
571 (0,0,0.3579,0.4168) (0.3579,0.4168,1,1)
765 (0,0,0.4869,0.5655) (0.4869,0.5655,0.6270,0.6286) (0.6270,0.6286,0.6293,0.6294) (0.6293,0.6294,0.6543,0.6769)
(0.6543,0.6769,0.7320,0.7667) (0.7320,0.7677,1,1)
802 (0,0,0.4227,0.7499) (0.4227,0.7499,1,1)
824 (0,0,0.6009,0.6017) (0.6009,0.6017,0.6026,0.6033) (0.6026,0.6033,1,1)
1168 (0,0,0.5665,0.5793) (0.5665,0.5793,1,1)
1346 (0,0,0.4839,0.5456) (0.4839,0.5456,1,1)
1423 (0,0,0.8269,0.8730) (0.8269,0.8730,1,1)
1546 (0,0,0.0792,0.3206) (0.0792,0.3206,0.4904,0.5156) (0.4904,0.5156,1,1)
1586 (0,0,0.9168,0.9753) (0.9168,0.9753,1,1)
1668 (0,0,0.2804,0.6472) (0.2804,0.6472,1,1)
1761 (0,0,0.5641,0.5764) (0.5641,0.5764,0.5784,0.5902) (0.5784,0.5902,1,1)
1772 (0,0,0.5156,0.5172) (0.5156,0.5172,1,1)
1867 (0,0,0.5292,0.6251) (0.5292,0.6251,1,1)
1939 (0,0,0.8908,0.8934) (0.8908,0.8934,1,1)
1990 (0,0,0.1022,0.3066) (0.1022,0.3066,0.4484,0.5811) (0.4484,0.5811,1,1)
Table 12: Features Partition in Leukemia dataset.
Fe.n. Partitions
2288 (0,0,0.0733,0.0835) (0.0733,0.0835,1,1)
2354 (0,0,0.1451,0.1931) (0.1451,0.1931,1,1)
3252 (0,0,0.0681,0.0706) (0.0681,0.0706,0.0738,0.0747) (0.0738,0.0747,1,1)
4644 (0,0,0.2425,0.2427) (0.2425,0.2427,1,1)
4847 (0,0,0.2116,0.2157) (0.2116,0.2157,0.3479,0.3531) (0.3479,0.3531,1,1)
6041 (0,0,0.1937,0.1963) (0.1937,0.1963,0.2001,0.2037) (0.2001,0.2037,1,1)
6376 (0,0,0.1408,0.1422) (0.1408,0.1422,1,1)
IJCCI2013-InternationalJointConferenceonComputationalIntelligence
330

Describing Selected Features
Table 13: Description of selected genes of the Colon dataset by FRF-fs method.
Imp. V. n gene Gene Description
35.6266 419 R44418 EBNA-2 Nuclear protein (Epstein-barr virus)
17.0359 765 M76378 Human cysteine-rich protein (CRP) gene, exons 5 and 6
15.6419 1635 M36634 Human vasoactive intestinal peptide (VIP) mRNA, complete cds
13.5216 824 Z49269 H.sapiens gene for chemokine HCC-1
13.4986 1168 U04953 Human isoleucyl-tRNA synthetase mRNA, complete cds
13.4898 513 M22382 Mitochondrial matrix protein P1 precursor (HUMAN)
9.63634 1772 H08393 Collagen alpha 2(XI) CHAIN (Homo sapiens)
7.23607 571 R42501 Inosine-5’-Monophosphate Dehydrogenase 2 (HUMAN)
7.04094 1546 T51493 Homo sapiens PP2A B56-gamma1 mRNA, 3’ end of cds
6.81338 1423 J02854 Myosin regulatory light chain 2, Smooth muscle isoform (HUMAN);
contains element TAR1 repetitive element
6.70853 1761 T94350 Peripheral myelin protein 22 (Homo sapiens)
6.60851 1939 X70297 Neuronal acetylcholine receptor protein, alpha-7 chain (HUMAN)
6.49896 1990 U15212 Human caudal-type homeobox protein (CDX1) mRNA, partial cds
5.99079 377 Z50753 H.sapiens mRNA for GCAP-II/uroguanylin precursor.
4.66543 1668 M82919 Human gamma amino butyric acid (GABAA) receptor beta-3 subunit mRNA,
complete cds.
4.09169 1346 T62947 60S RIBOSOMAL PROTEIN L24 (Arabidopsis thaliana)
3.19286 1586 L14848 Human MHC class I-related protein mRNA, complete cds.
2.37430 548 T40645 Human Wiskott-Aldrich syndrome (WAS) mRNA, complete cds.
2.01753 474 T70046 Endothelial actin-binding protein (Homo sapiens)
1.83728 802 X70326 H.sapiens MacMarcks mRNA
1.73155 1867 U32519 Human GAP SH3 binding protein mRNA, complete cds.
1.71548 1724 H16991 Nucleolysin tiar (HUMAN)
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Table 14: Description of selected genes of the Leukemia dataset by FRF-fs method.
Imp. V. n gene Gene Description
31.2849 3252 U46499 Glutathione S-transferase, Microsomal
30.1804 1882 M27891 CST3 Cystatin C (amyloid angiopathy and cerebral hemorrhage)
30.1763 1834 M23197 CD33 CD33 antigen (differentiation antigen)
26.5833 4847 X95735 Zyxin
23.9430 2288 M84526 DF D component of complement (adipsin)
13.5707 2354 M92287 CCND3 Cyclin D3
13.1465 6041 L09209 s APLP2 Amyloid beta (A4) precursor-like protein 2
9.87071 6376 M83652 s PFC Properdin P factor, complement
4.86655 4644 X80230 mRNA (clone C-2k) mRNA for serine/threonine protein kinase
1.4004 3623 U68727 Homeobox-containing protein mRNA
1.2354 4708 X84002 TAFII20 mRNA for transcription factor TFIID
1.1158 5691 D89377 Adult tooth pulp of third molar fibroblast mRNA for MSX-2
0.9525 6855 M31523 TCF3 Transcription factor 3 (E2A immunoglobulin enhancer binding factors
E12/E47)
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
UsingaFuzzyDecisionTreeEnsembleforTumorClassificationfromGeneExpressionData
331
