
Toxic Mechanisms of α-Amanitin and Its Potential to Fight Cancer
Xueke Bai
1,
†
a
, Peixi Jiang
2,
†
b
and Yuyou Wu
3,
†
c
1
Department of Chemistry, The University of Manchester, Manchester, U.K.
2
Department of Applied Chemistry, Central South University, Changsha, Hunan, China
3
Faculty of Arts and Science, University of Toronto, Toronto, ON, Canada
†
These authors contributed equally
Keywords: α-Amanitin, Cancer Treatment, Selective RNA Inhibition.
Abstract: In fighting one of the major health problems in the world, the common approach to cancer treatment is the
combination of chemotherapy and radiation therapy yet from which the cytotoxic effects on normal tissues
and the drug tolerance gained through remain a huge obstacle ahead; thus, new approaches are in immediate
demand. In the past decades, toxins such as α-Amanitin have been studied in treating colorectal cancer, breast
cancer and pancreatic tumor as a therapeutic agent mostly by conjugating to moieties with targeting property
and reduced toxicity. The Amanitin toxin blocks RNA polymerase II activity and is the most specific and
most potent inhibitor of the eukaryotic transcription, hepatotoxicity being the main syndrome. For the
selective inhibition of RNAPII and induced RNPII activity in cancer cells, using this transcriptional arrest to
fight cancer cells appears to be a novel approach with broad applications. Considering the liver toxicity of α-
Amanitin, conjugation of α-Amanitin to antibodies or small molecules minimizes its toxicity and increases
the efficacy of treatment with relatively accurate targeting properties. In addition, further enhancements such
as photocaged α-Amanitin analog, conjugation with pH low insert peptide (pHLIP) and Fc Domain provide
access to more controlled drug release and ideal pharmacokinetics.
1 INTRODUCTION
Cancer is a major health problem across the world;
from the statistics of the National Health Center, 28.4
million of new cancer cases are expected by 2040
Sung. Cancer is a disease caused by certain genes
changes; TP53 is such a well-known tumor
suppressor gene and is frequently inactivated by a
deletion in a majority of human tumors. Cancer cells
grow uncontrollably and invade other parts of the
body, instead of dying through a process known as
programmed cell death as normal cells do.
Additionally, cancer relapse after chemotherapy is a
frequent biological phenomenon, along with survival
of the subpopulations drug-tolerant colonies (DTCs).
While common cytotoxic therapies are detrimental to
normal tissues, cancer cells can also develop
resistance to chemotherapy. Consequently, new
approaches are urgently required for successful
a
http://orcid.org/0000-0003-0058-4432
b
https://orcid.org/0000-0002-7694-1048
c
http://orcid.org/000-0003-4420-2248
administration in humans where the inhibitory effect
is strongly selective thus reducing adverse effects on
normal tissues. Recent studies have found the
enhanced antitumor activity of monoclonal
antibodies by conjugation to cytotoxic agents
(Shuptrine, 2012). Combining the highly-selective
property of antibody and the cytotoxic molecules
(payloads) with a linker providing covalent binding,
Antibody-Drug Conjugates (ADC) make it possible
for this specific route where the toxin is delivered to
kill target cells avoiding normal tissues; several of
these them are currently being evaluated in clinical
trials against cancer (Strassz, 2020). Except for
ADCs, Small Molecule-Drug Conjugates (SMDC)
have also been designed to overcome limitations in
ADCs. Following specific approaches to targets, the
conjugates readily enter tumor cells and release active
payloads for further inhibition. So far ADCs are
based on only a few toxic compounds, which are
Bai, X., Jiang, P. and Wu, Y.
Toxic Mechanisms of -Amanitin and Its Potential to Fight Cancer.
DOI: 10.5220/0011378000003443
In Proceedings of the 4th International Conference on Biomedical Engineering and Bioinformatics (ICBEB 2022), pages 1095-1103
ISBN: 978-989-758-595-1
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
1095

largely limited to microtubule- or DNA-targeting
toxins that mainly impact proliferating cells and have
limited efficacy in diseases with a low proliferative
fraction such as indolent lymphomas or multiple
myeloma (Strassz, 2020). This limitation further
urges the studies of new compounds with alternative
toxic mechanisms and the ability to selectively inhibit
cancer cells.
Amatoxins can be found in several species of the
mushroom genus Amanita, one being the famous
death cap (Amanita Phalloides), and also in the
mushrooms Galerina marginate and Conocybe filaris.
Among the amatoxin family, α-Amanitin is possibly
the most fatal one. The toxin is notorious for its
specific and non-covalent binding to RNA
polymerase II, thereby decreasing mRNA levels and
protein synthesis, which is the primary toxic
mechanism for liver necrosis or apoptosis (Arima,
2005, Leu and George, 2007, Ljungman, 1999).
Among the twelve subunits in the human RNA
polymerase II complex, POLR2A encodes the largest
subunit that is indispensable for the polymerase
activity in mRNA synthesis. In addition, genomic
deletion of the tumor suppressor gene TP53
frequently comes with encompassment of
neighboring essential gene POLR2A, rendering
cancer cells with hemizygous TP53 deletion
vulnerable to further suppression of POLR2A
expression, which can be inhibited by α-Amanitin
(Liu, 2015). Molecular events that cause tumor
formation involve a number of Homeobox (Hox)
genes, proteins of which can bind to regulatory DNA
at the level of transcription of target gene DNA to
messenger RNA by RNA polymerase II (Boube,
2014). Comparing with other toxins employed in
ADCs development such as microtubule inhibitors
and DNA cross-linkers, α-Amanitin ADCs have an
effect on slowly dividing tumor cells (Hechler, 2014).
Additionally, considering the number of intracellular
targets of ADCs, RNAPII are fewer than targets of
other developments, which leads to a lower
concentration of α-Amanitin having complete
inhibition (Rudd and Luse, 1996). Thus, the selective
inhibition of RNAPII plays a crucial role in the
inhibitory effect against cancer.
This review discusses the recent research on α-
Amanitin as an RNAPII inhibitor with a focus on its
high selectivity and controlled drug pathway that
allows new innovative and effective therapeutics
against cancer. The chemical properties of α-
Amanitin and its activity in cancer cells are also
discussed in this review. As the non-stop mRNA
synthesis and gene expression are essential in the
endless growth of cancer cells, and studies have
demonstrated the mechanism underlying relapse is
based on transcriptional regulation, α-Amanitin
presents itself as a novel therapeutic agent against
human cancer (Kume, 2016). Although applications
of free α-Amanitin are limited in use due to its liver
toxicity, α-Amanitin-based conjugates were
evaluated with reduced toxicity for its potential to
suppress DTC formation and reduce cancer cells
resistance. The conjugates appeared optimistic in
vivo. While the large size of ADCs impairs the
penetration and SMDCs have better tumor-
penetrating properties, the pharmacokinetics of
SMDCs limit their anti-tumor activity. Consequently,
more enhancements of these approaches are
discussed to overcome the limitations; photocaged
Amanitin analogs are introduced as a novel method
to control drug release (Matinkhoo, 2021);
conjugation with an Fc Domain enhances the
pharmacokinetics and anti-tumor activity of α-
Amanitin-based-SMDCs (Gallo, 2021).
2 PROPERTIES OF AMANITINS
α-Amanitin is a highly toxic bicyclic octapeptide with
eight amino acids and is produced by the hepatotoxic
mushroom genus Amanita with its relatives β-, γ-,
and ε-Amanitin. The Amanitins are special peptides
with the amino acid chains branched and the branches
giving rise to an inner loop. The structural derivatives
of α-Amanitin show the importance of bridge helix
interaction for inhibitory activity (Fig.1a) (Wang,
2011, ZANOTTI, 1987). α-Amanitin is possibly
considered to be the deadliest among all the
amatoxins by far. The mushroom species which
called Amanita Phalloides ("death cap") or Amanita
verna (“destroying angel”) is famous for its red cap
and lethal toxin Amanitin. It has been estimated that
Amanita Phalloides is responsible for 90% of the
mushroom-ingested fatalities worldwide with
hepatocellular failure being the main syndrome (α-
Amanitin poisoning may require liver transplantation
when the toxicity is severe). Chemical and physical
properties of α-Amanitin were shown in Table 1.
Amatoxins can be absorbed rapidly after ingestion.
The ingested amounts as low as 0.1 mg/kg are
sufficient to be lethal (Lewis and Seeff 2020). The
oral LD₅₀ of Amanitin is 0.4-0.8 mg/kg in mice and
it takes nearly 2–8 days to cause death (Saravanapriya
and Devi 2021).
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1096
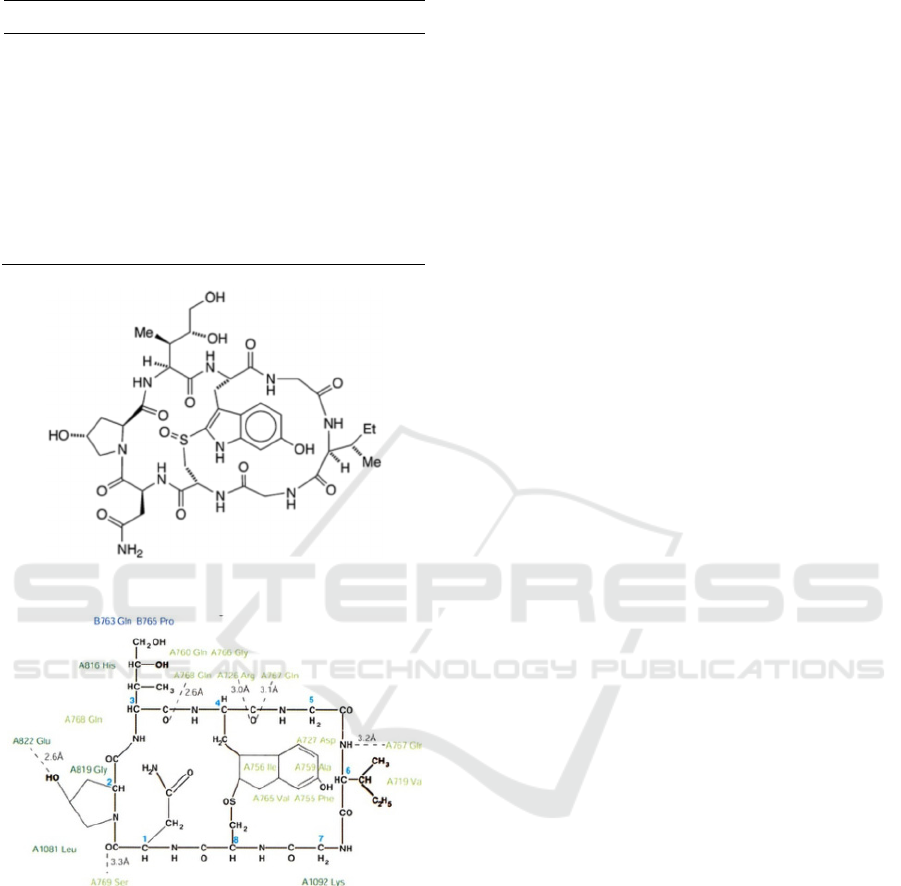
Table 1: Chemical and physical properties of α-Amanitin.
Pro
p
ert
y
Name Pro
p
ert
y
Value
Em
p
irical Formula C
39
H
54
N
10
O
14
S
Average Molar Mass 918.97 g/mol
H
y
dro
g
en Bond Donor Count 13
Hydrogen Bond Acceptor Count 15
Rotatable Bond Count 7
Melting Point 254-255 ºC
Water Solubility
ൎ1-10 g/L
(a) The bicyclic octapeptide amatoxins
(b) Interaction of α-amanitin with RNAPII
Figure 1: Chemical structure of α-Amanitin with residues
of with RNAPII (Bushnell, 2002).
3 MOLECULAR TOXICITY
MECHANISMS OF ALPHA-
AMANITIN
Amatoxins can block nuclear RNA polymerases via
the target organs (intestinal mucosa, liver and
kidneys) to inhibit synthesis of proteins. Liver is the
privileged target of α-Amanitin (Amatoxins can be
absorbed by the gastrointestinal tract and then travel
within the enterohepatic circulation and reach the
hepatocytes, where the inhibition of mRNA
production and protein synthesis occurred, causing
cell necrosis, inducing apoptosis and glutathione
depletion), internalizing the toxin through the
organic-anion transporter 1B3 (OATP1B3)
(Letschert, 2006), and thus receiving a massive
amount of Amanitin after rapid gastrointestinal
absorption.
3.1 Pro-apoptosis Activity
Inside the cell, α-Amanitin induces the stress signals
(ex. RNA polymerase inhibition) which lead to an
induction of the p53 protein, allowing the formation
of complexes with the two anti-apoptotic proteins
which called B-cell lymphoma-extra-large (Bcl-XL)
and B-cell lymphoma-2 (Bcl-2) and triggering
apoptosis by the mitochondrial release of cytochrome
c in the cytosol (Arima 2005, Leu and George 2007,
Ljungman 1999). F.A. Derheimer et al. presented two
possible mechanisms of tumor suppressor gene p53
induction by α-Amanitin (Derheimer 2007). The
study suggested that the export of p53 from the
nucleus is dependent on the export of mRNA so that
when the synthesis or export of mRNA is blocked,
p53 accumulates in the nucleus by default. Secondly,
inhibition of transcription elongation leads to the
phosphorylation of the Ser-15 site of p53. Blockage
of transcription is sufficient for the nuclear
accumulation of p53 even though it's unclear that
which mechanism takes place in the cells. A report
pointed out that α-Amanitin induced significant
changes in the mitochondrial proteome, associated
with the destruction of membrane potential (Wang
2018). Hence, Amanitin-induced apoptosis has been
considered to play a vital role in the pathophysiology
of these intoxications (Figure 2) (Magdalan, 2010).
3.2 Enhancement of Oxidate Stress
Some mechanisms have been suggested the
formation of reactive oxygen species (ROS) leading
to oxidative stress-related damages. The production
of oxidative stress has been seen as an essential factor
in the development of that severe hepatotoxicity. In
fact, some studies have shown that the accumulation
of α-Amanitin leads to the increase of superoxide
dismutase (SOD) and glutathione (GSH) peroxidase
activities, malondialdehyde products, and lipid
peroxidation, which is related to the inhibition of
catalase activity (Dündar, 2017, Rodrigues, 2020,
Toxic Mechanisms of -Amanitin and Its Potential to Fight Cancer
1097

Steurer, 2018, Zheleva, 2013, Zheleva, 2007) (Figure
2). Recent researches have indicated that α-Amanitin
induces the production of GSH and tGSH, confirming
the hypothesis of the involvement of oxidative stress
in the pathophysiology (Rodrigues 2020, Steurer
2018). A scientist found that α-Amanitin could form
phenoxyl-free radicals that might be involved in the
increased production of reactive oxygen species
(Figure 2) (Zheleva, 2013). With the case of
Amanitin intoxication, induction of the NF-κB
(nuclear factor-kappa B) pathway has been observed
to have a certain protective effect without
establishing a link with the levels of production of
SOD, GSH, or catalase (Garcia, 2015, Morgan and
Liu, 2011).
3.3 Inhibition of Protein Synthesis
Furthermore, the cytotoxicity α-Amanitin will result
in the inhibition of RNA polymerase, especially RNA
polymerase II (it is more sensitive to this mushroom
toxin than other polymerases in eukaryotic cells).
Since RNA polymerase II is responsible for mRNA
synthesis in the cell, α-Amanitin is a potent and
selective inhibitor of mRNA synthesis (Kume, 2016).
The main toxicity mechanism of Amanitin is
attributed to non-covalent nuclear inhibition of RNA
polymerase type II (RNAP II), which reduces mRNA
levels and protein synthesis (WIELAND 1983). By
complexing with the intracellular RNA polymerase II
enzyme, amatoxins inhibit the formation of mRNA
and then restrain the protein synthesis, leading to cell
necrosis rapidly. This enzyme inhibition has been
proved to be the cause of RNAP II ubiquitination and
its degradation by proteasomes, which is related to
the increased intracellular ATP concentration
(Rodrigues, 2020, Steurer, 2018) (Figure 2). Up to
now, there are still many unknown areas about the
toxic effects of Amanitin at the cellular level that
need further exploration. The treatment mainly
remains symptomatic.
Figure 2: Main toxicity mechanisms of Amanitin within
hepatocytes (modified from (Le Daré, 2021)
4 INTRACELLULAR
ACTIVITIES OF
ALPHA-AMANITIN
4.1 Inhibition of Tumor Growth
through Suppression of POLR2A
While there is no denying that most cancers come
along with the deletion of the tumor suppressor gene
p53, no effective p53-based therapy has been
successfully applied in clinical treatment due to its
complexity in signalling. However, 104 (53%) out of
195 colorectal cancer cells (CRC) cases bear the
hemizygous loss in the 17p13 region, resulting in
concomitant deletion of TP53 and POLR2A(Liu,
2015). The gene POLR2A encodes the largest subunit
of RNA polymerase II and is indispensable in cell
proliferation. No homozygous deletion was observed
in cancer cells, in accordance with the fact that
POLR2A is essential for cancer cell survival. Studies
demonstrated that POLR2A expression tightly
correlates with its gene copy numbers, resulting in
significantly lower levels of POLR2A loss
(hemizygous deletion) cells(Liu, 2015). However, by
comparing POLR2Aneutral cells and POLR2Aloss
cells, the similar proliferation rates indicate this
hemizygous loss is sufficient to main proliferation in
HCT116 cells. The half-maximum inhibitory
concentration (IC50) for the POLR2Aneutral cells
was 10-fold greater than the POLR2Aloss cells; when
examined for drug sensitivity to different
chemotherapy drugs of these two cell types, POLR2A
inhibited by α-Amanitin notably increased the cell-
killing effects yet no significant enhancements were
observed in normal cells. POLR2Aloss cells were
more sensitive to POLR2A inhibition by α-Amanitin
while re-expression of POLR2A rescued resistance to
α-Amanitin, where this loss happens to be concurrent
with p53 deletion in major cancer cases. High
concentrations of α-Amanitin caused complete deaths
while with low doses, the inhibition had significantly
higher levels of cell-killing effect on the POLR2A
loss cells than the neutral ones(Liu 2015). Taken
together, this research implied using α-Amanitin as a
potential therapeutic method against CRC.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1098

4.2 Inhibition of RNAPII by Amanitin
via TAF15 Mrna
Κume et al. investigated the impact of RNA
polymerase II inhibition on DTC survival(Kume,
2016). Among four compounds that affect different
steps including chromatin formation, transcription, or
protein synthesis, α-Amanitin showed remarkable
suppression of colony formation in the shortest
exposure period. Moreover, colony formation was
also clearly suppressed by the other less-specific
RNAP inhibitor AMD, suggesting the selective
inhibition of RNAPII plays a crucial role in
suppressing colony formation. Their study also
demonstrated that TAF15 is especially responsible
for DTC formation as TAF15 gene products are
upregulated in DTC-forming cells. Recent studies
had suggested TAF15 binds the C-terminal domain of
RNAPII more avidly than other RNA-binding
proteins in the TET family and acts as a coactivator
of RNAPII(Kwon, 2013). Both TAF15 mRNA and
protein levels were decreased after treatment,
suggesting RNAPII activity towards TAF15 mRNA
was inhibited by α-Amanitin. Furthermore, the
subsequent colony formation assay showed that
TAF15 knockdown suppressed the emergence of
both DTCs with or without α-Amanitin treatment,
meaning TAF15 depletion inhibited DTC formation.
Additionally, TAF15-knockdown and α-Amanitin-
treated cells induced similar morphological changes,
suggesting a TAF15 depletion by α-Amanitin
treatment. Both TAF15 mRNA and protein levels
were decreased in response to α-Amanitin, consistent
with similar morphological changes. These results
suggested TAF15 is a mediator of RNA polymerase
II-dependent DTC formation and a crucial target of
α-Amanitin. The RNAPII-dependent inhibition of
early-phase mRNA synthesis is sufficient to produce
a nearly complete suppression of colony formation,
and the practicality of α-Amanitin treatment for
disease caused by DTCs, such as Peritonitis
Carcinomatosa by chemotherapy, via inhibition of
TAF15.
5 THERAPEUTIC USE OF
ALPHA-AMANITIN
α--Amanitin has a low cellular uptake due to its polar
structure and low permeation except in liver cells
where OATP1B3 internalizes the toxin(Bodero
2018). Free Amanitin permeation mediated by
transporter protein results in apoptosis and necrosis
of hepatocytes(Bodero, 2018, Letschert, 2006). The
strong inhibition effect caused by Amanitin aroused
the interest of scientists to target cancer cells while
preventing liver toxicity. Numerous studies have
shown promising cell-killing effects after direct α-
Amanitin treatment in tumor cells. In order to
enhance the selectivity of Amanitin treatment,
scientists have developed conjugates targeting tumor
cells and novel drug release methods.
5.1 Antibody-drug Conjugates
Approach
Amanitin antibody-drug conjugates (ADC) were
found to successfully increase α-Amanitin activity in
target cells while not being a substrate of OATP1B3
to avoid systematic toxicity. The antibody is
responsible for targeting specific antigens and
bringing the toxin α-Amanitin into the cells.
Moreover, epithelial cell adhesion molecule
(EpCAM) is a target antigen highly and frequently
expressed on carcinoma cells and its metastasis
(Gires 2020). α-Amanitin that was conjugated with
chiHEA125, a chimerized anti-EpCAM antibody,
reduced cell proliferation in human pancreatic
(BxPc-3 and Capan-1), colorectal (Colo205), breast
(MCF-7), and bile duct (OZ) cancer cell lines (IC50
= 2.5×10−10 to 5.4×10−12 M)(Moldenhauer 2012).
The antiproliferative effect of chiHEA125-ama was
up to 10,000 -fold higher than α-Amanitin alone.
Anti-tumor effects of chiHEA125-Amanitin
conjugates were tested in an experimental human
BxPc-3 pancreatic cancer model, induced by
injecting BxPc-3 cancer cells subcutaneously into the
right flank of female mice. A single dose (50 μg/kg
with respect to α-Amanitin) suppressed BxPc-3
xenograft tumor growth, while two higher doses (100
μg/kg with respect to α-Amanitin), administered one
week apart, inhibited tumor recurrence for 3-4 weeks.
Besides, the treatment was well-tolerated in tumor-
bearing mice and no substantial difference was
observed compared with the control mice treated with
unconjugated chiHEA125 (Moldenhauer, 2012).
Drug resistance is one of the major concerns of
existing chemo- and targeted therapies of colorectal
cancer cells (CRC)(Van der Jeught, 2018). Escaping
mechanisms including enhanced DNA repair and
drug metabolism result in worse clinical outcomes.
Research showed that α-Amanitin-HEA125
selectively killed human CRCs with a hemizygous
deletion of POLR2A(Liu 2015). Those cells were
more sensitized to chemotherapy drugs owing to the
strong inhibition of POLR2A by α-Amanitin. It was
observed that a very low dose of α-Amanitin-
Toxic Mechanisms of -Amanitin and Its Potential to Fight Cancer
1099

HEA125 (10μg/kg) was sufficient to inhibit tumor
growth in mice bearing POLR2Aloss HCT116
tumors, reducing the effective doses of α-Amanitin
by at least 10,000-fold (IC50 = 0.01 ng/ml).
Increasing specificity and effects on chemo-resistant
cancer cells by conjugating with antibody HEA125
implied a potential therapeutic way to EpCAM
expressed human cancers and other Amanitin ADCs
might be synthesized in future studies to target other
cancers. Although α-Amanitin ADC has shown
positive outcomes in various animal studies, this
approach is relatively high cost and may result in
unfavorable pharmacokinetics and immune response
(Bodero, 2018).
Antibody-targeting Amanitin conjugates
(ATACs) have a broader application than other toxin
compounds as payloads because of their unique
interaction with RNAPII. Comparing with other
intracellular targets like microtubule(Nasiri 2018),
RNAPII maintains a much lower number of 100 to
1000, rendering low concentration able to achieve
optimistic cell-killing effects. Slow growth of tumor
is common in prostate cancers, and another advantage
of α-Amanitin as a payload is the active inhibition on
dormant cells. Despite the fact that Prostate-specific
membrane antigen (PSMA) are expressed in normal
tissues as well, the expression levels are much higher
in prostate carcinomas, indicating a potential high
specificity for antibody therapy(Osborne 2013).
Scientists conjugated α-Amanitin to the anti-PSMA
mAb 3F11(Hechler, 2014). To test the active
inhibition of ATACs on non-proliferating cells,
growth arrest of LNCaP cells is made by addition of
Interleukin 6 (IL-6). Inhibition effects were observed
both on rapidly dividing and growth arrested LNCaP
cells. The viability curve of dormant cells is
comparable to dividing ones under increasing
concentration of Amanitin ADC, indicating the
proliferation-independent inhibition on tumor by
ATACs (Hechler, 2014).
However, the large size and high affinity to the
target of ADCs restrict their ability of penetration,
especially in solid tumors. Furthermore, their long
circulatory half-life might lead to immunogenicity
and unspecific toxicities. Thus, alternative
approaches may still catch scientists’ attention in the
future.
5.2 Small Molecule-drug Conjugates
Approach
Small molecule-drug conjugates (SMDCs) are an
alternative approach where a specific cell-membrane-
receptor ligand assists drug delivery to the target site
and internalization by the receptor. Tripeptide
arginine-glycine-aspartate (RGD) and the related
sequence isoaspartate-glycine-arginine (isoDGR)
were two ligands that have been conjugated to α-
Amanitin targeting the αVβ integrin receptor family
(Bodero 2018). αVβ integrin receptors are strongly
expressed on blood vessels in human cancers such as
breast cancer, glioblastoma, pancreatic tumor,
prostate carcinoma (Desgrosellier and Cheresh
2010). The conjugates demonstrated great binding
affinity to the receptor like the free ligands. However,
the toxicity and inhibitory effect of the integrin
ligand-α-Amanitin conjugates were either worse or
slightly better than free α-Amanitin. Similar results
were achieved in other studies involving small
molecule-amanitin conjugates (Moshnikova, 2013,
Zhao, 2015). In addition, SMDCs have a shorter half-
life compared to ADCs due to their smaller size,
limiting their distribution to tumor cells and
therapeutic effect. In order to prolong circulatory
half-life, the immunoglobulin Fc domain was
conjugated to α-Amanitin-based SMDCs (Gallo,
2021). Apparently, the interaction between the Fc
domain and neonatal Fc receptors (FcRN) results in
prolonged exposure to drugs that contain Fc peptides
(Wang, 2011, Wu, 2012). Both SMDCs (IC50 =
0.863 nM)) and Fc-SMDCs (IC50 = 15.2 nM) had
higher cytotoxicity in vitro compared to
unconjugated α-Amanitin (IC50 = 476 nM). In vivo
pharmacokinetics and biodistribution study were
conducted to evaluate half-live, tumor, and organ
accumulation of SMDCs and Fc-SMDCs. Fc-SMDCs
showed a dramatic decrease in their clearance, thus
extending their half-lives from 44 min to
approximately 7.2 days (Gallo, 2021).
Table.2: Evaluation of inhibitory effect of α-Amanitin and α-Amanitin conjugates in various cell lines.
Compound (name) IC
50
(cell lines) References
ChiHEA125-Ama 2 × 10
−12
M (Colorectal,
Colo205), 8.7 × 10
−11
M (Bile
duct, OZ), 2.1 × 10
−11
M
(Pancreatic, Capan-1), 2.5 × 10
−10
M (Pancreatic, BxPC-3), 5.4 × 10
−12
M (Breast, MCF-7)
(Moldenhauer 2012)
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1100

Ama-HEA125 0.1 μg/ml (POLR2A
loss
HCT116) (Liu 2015)
HDP 30.2284 8.63 × 10
-10
M
(
LNCaP
)
(
Gallo 2021
)
HDP 30.2972 1.52 × 10
-8
M (LNCaP) (Gallo 2021)
α-Amanitin ~4.76 × 10
-8
M
(
Gallo 2021
)
To increase the cytotoxicity effect, α-Amanitin
was combined with another cytotoxic drug displaying
an independent mode of action. Dual conjugation of
α-Amanitin and monomethyl auristatin E (MMAE)
with fibroblast growth factor 2 (FGF2) tend to
increase the toxicity of both drugs and maintain the
selectivity (Świderska, 2018). Fluorescence
microscopy was performed to track the
internalization process and found that FGF2 dual
conjugate internalization was strongly correlated with
FGF receptors level on the cell surface. None of the
tested conjugates displayed severe toxicity towards
receptor-negative cells, suggesting its selectivity to
FGF receptor expressed cells. After binding to the
high affinity FGFRs on the cancer cell surface, dual
FGF2 conjugate is internalizing by endocytosis.
Processing through the endosome–lysosome pathway
leads to release of MMAE and α-Amanitin inside the
cell and next respectively inhibit tubulin
polymerization and DNA transcription. α-Amanitin
inhibited DNA transcription and MMAE stopped
microtubule polymerization simultaneously, leading
to cell apoptosis. The dual conjugate had a greater
cytotoxic effect than any of single-drug FGF2
conjugates, which can be explained as a result of the
combined cytotoxic action of α-Amanitin and
MMAE. With the highest concentration, α-
Amanitin/MMAE-FGF2 conjugate reduced 95% of
cell viability. No or minimal immune response was
triggered since the ligand sequence was fully from
Homo sapiens (Świderska, 2018). The positive result
of combining two cytotoxic agents allows more
possibilities in the future against different cancer cells
with different intracellular impair.
5.3 Drug Delivery and Release
On the basis of cell targeting models including ADCs
and SMDCs, controlled drug release further increases
selectivity. Stimuli-responsive prodrugs and carriers
allowed on-demand drug delivery and release
(Dunkel and Ilaš, 2021). Photocaged α-Amanitin
analog, Ama-Flash has been synthesized by
Matinkhoo and coworkers (Matinkhoo 2021).
Nitroveratryl (Nv) ether was attached to modified α-
Amanitin as a photo-masking group which is
extensively used in photo-pharmacology studies. Nv-
protected α-Amanitin was inactive against RNAP II
and didn't result in cell death even at a high dose of
100 μM. α-Amanitin analog was released through a
25 min irradiation with λ = 366 nm and CHO
(Chinese hamster ovary) cell viability was measured
after 48 and 72 hours, with α-Amanitin as a control.
The IC50 value was not significantly different from
free α-Amanitin, suggesting that irradiation may not
affect toxicity. Furthermore, irradiated wavelengths
are minimal cytotoxic and won't be absorbed by
tryptathionine in α-Amanitin. The toxicity of
byproduct 4,5-dimethoxy-2-nitrobenzyl (DMNB)
was not tested. With light-activated α-Amanitin
analog, it creates the possibility to preload the drug
into cells and trigger RNAP II inhibition and
ubiquitination when needed. Amanitin conjugated
with pH low insert peptide (pHLIP) is another drug
delivery method based on pH-dependent
conformational change of pHLIP which leads to its
insertion into the membrane (Moshnikova, 2013).
Acidic environment protonates Asp/Glu residues and
increases hydrophobicity of pHLIP. Studies showed
that the antiproliferative effect was 4-5 times higher
at pH 6 compared to pH 7.4, creating the selectivity
due to the negative transmembrane pH gradient of
cancer cells. Cytotoxic effect was achieved on four
different human cancer cell lines at concentration of
0.25–1 μM. Taken together, the new techniques of
improving drug delivery and release such as ADCs,
SMDCs, as well as photo-pharmacology methods,
give more flexibility to cancer chemotherapy.
6 CONCLUSIONS
In this review, the toxic mechanisms of α-Amanitin,
a fetal chemical present in the mushroom species
Amanita Phalloides were discussed. Its ability to
inhibit mRNA polymerase II and induce p53 protein
makes it a new possibility against cancer. We
reviewed multiple animal studies in which α-
Amanitin conjugates were formed with either
antibodies or small molecules and showed strong
selectivity and toxic effect. Novel drug delivery
methods provide new insight into future cancer
chemotherapy. More in vivo and in vitro animal
studies with larger sample sizes are necessary in order
Toxic Mechanisms of -Amanitin and Its Potential to Fight Cancer
1101

to confirm the cytotoxicity effect as some cytotoxic
mechanisms are still unclear. There are risks
associated with animal-to-human extrapolation due to
differences in metabolism and size. A first-in-human
phase 1/2a study started enrolling participates in early
2021 (Strassz, 2020). and this study aims to
determine the maximum tolerated dose and assess the
anti-tumor activity of HDP101, an ADC targeting
BCMA (B cell maturation antigen) carrying a
synthetic version of Amanitin as a payload. Future
research focusing on the mechanisms of α-Amanitin
anti-cancer effects and related clinical trials may be
required to promote the understanding of α-Amanitin
as a potential therapeutic way for cancer treatment.
REFERENCES
Arima, Y., M. Nitta, S. Kuninaka, D. Zhang, T. Fujiwara,
Y. Taya, M. Nakao, and H. Saya. (2005)
Transcriptional blockade induces p53-dependent
apoptosis associated with translocation of p53 to
mitochondria. Journal of Biological Chemistry,
280:19166-19176.
Bodero, L., P. López Rivas, B. Korsak, T. Hechler, A. Pahl,
C. Müller, D. Arosio, L. Pignataro, C. Gennari, and U.
Piarulli. (2018) Synthesis and biological evaluation of
rgd and isodgr peptidomimetic-α-amanitin conjugates
for tumor-targeting. Beilstein Journal of Organic
Chemistry, 14:407-415.
Boube, M., B. Hudry, C. Immarigeon, Y. Carrier, S.
Bernat-Fabre, S. Merabet, Y. Graba, H.-M. Bourbon,
and D.L. Cribbs. (2014) Drosophila melanogaster hox
transcription factors access the rna polymerase ii
machinery through direct homeodomain binding to a
conserved motif of mediator subunit med19. PLoS
Genetics, 10:e1004303.
Bushnell, D.A., P. Cramer, and R.D. Kornberg. (2002)
Structural basis of transcription: -amanitin-rna
polymerase ii cocrystal at 2.8 a resolution. Proceedings
of the National Academy of Sciences, 99:1218-1222.
Derheimer, F.A., H.M. O'Hagan, H.M. Krueger, S.
Hanasoge, M.T. Paulsen, and M. Ljungman. (2007)
Rpa and atr link transcriptional stress to p53.
Proceedings of the National Academy of Sciences,
104:12778-12783.
Desgrosellier, J.S. and D.A. Cheresh. (2010) Integrins in
cancer: Biological implications and therapeutic
opportunities. Nature Reviews Cancer, 10:9-22.
Dündar, Z.D.E., M.; Kilinç, I.; Çolak, T.; Oltulu, P.;
Cander, B. (2017) Dündar, z. D., ergin, m., kilinç, i.,
çolak, t., oltulu, p., cander, b. (2017). The role of
oxidative stress in α-amanitin-induced hepatotoxicityin
an experimental mouse model. Turkish Journal of
Medical Sciences, 47:318–325.
Dunkel, P. and J. Ilaš. (2021) Targeted cancer therapy using
compounds activated by light. Cancers, 13:3237.
Gallo, F., B. Korsak, C. Müller, T. Hechler, D. Yanakieva,
O. Avrutina, H. Kolmar, and A. Pahl. (2021) Enhancing
the pharmacokinetics and antitumor activity of an α-
amanitin-based small-molecule drug conjugate via
conjugation with an fc domain. Journal of Medicinal
Chemistry, 64:4117-4129.
Garcia, J., V.M. Costa, A.T.P. Carvalho, R. Silvestre, J.A.
Duarte, D.F.A.R. Dourado, M.D. Arbo, T. Baltazar,
R.J. Dinis-Oliveira, P. Baptista, M. de Lourdes Bastos,
et al. (2015) A breakthrough on amanita phalloides
poisoning: An effective antidotal effect by polymyxin
b. Archives of Toxicology, 89:2305-2323.
Gires, O., M. Pan, H. Schinke, M. Canis, and P.A. Baeuerle.
(2020) Expression and function of epithelial cell
adhesion molecule epcam: Where are we after 40
years? Cancer metastasis reviews, 39:969-987.
Hechler, T., Kulke, M., Müller, C., Pahl, A. and Anderl, J.
(2014) Poster presentation #664: amanitin-based
antibody-drug conjugates targeting the prostate-
specific membrane antigen psma. AACR Annual
Meeting.
Kume, K., M. Ikeda, S. Miura, K. Ito, K.A. Sato, Y.
Ohmori, F. Endo, H. Katagiri, K. Ishida, C. Ito, T.
Iwaya, et al. (2016) Α-amanitin restrains cancer relapse
from drug-tolerant cell subpopulations via taf15.
Scientific Reports, 6:25895.
Kwon, I., M. Kato, S. Xiang, L. Wu, P. Theodoropoulos, H.
Mirzaei, T. Han, S. Xie, J.L. Corden, and S.L.
McKnight. (2013) Phosphorylation-regulated binding
of rna polymerase ii to fibrous polymers of low-
complexity domains. Cell, 155:1049-1060.
Le Daré, B., P.-J. Ferron, and T. Gicquel. (2021) Toxic
effects of amanitins: Repurposing toxicities toward
new therapeutics. Toxins, 13.
Letschert, K., H. Faulstich, D. Keller, and D. Keppler.
(2006) Molecular characterization and inhibition of
amanitin uptake into human hepatocytes. Toxicological
Sciences, 91:140-149.
Leu, J.I.J. and D.L. George. (2007) Hepatic igfbp1 is a
prosurvival factor that binds to bak, protects the liver
from apoptosis, and antagonizes the proapoptotic
actions of p53 at mitochondria. Genes & Development,
21:3095-3109.
Lewis, J.H. and L.B. Seeff. (2020) The origins of the
modern-day study of drug hepatotoxicity: Focus on
hyman j. Zimmerman. Clinical liver disease, 15:S25-
S36.
Liu, Y., X. Zhang, C. Han, G. Wan, X. Huang, C. Ivan, D.
Jiang, C. Rodriguez-Aguayo, G. Lopez-Berestein, P.H.
Rao, D.M. Maru, et al. (2015) Tp53 loss creates
therapeutic vulnerability in colorectal cancer. Nature,
520:697-701.
Ljungman, M., F. Zhang, F. Chen, A.J. Rainbow, and B.C.
McKay. (1999) Inhibition of rna polymerase ii as a
trigger for the p53 response. Oncogene, 18:583-592.
Magdalan, J., A. Ostrowska, A. Piotrowska, I. Izykowska,
M. Nowak, A. Gomułkiewicz, M. Podhorska-Okołów,
A. Szelag, and P. Dziegiel. (2010) Alpha-amanitin
induced apoptosis in primary cultured dog hepatocytes.
Folia Histochemica et Cytobiologica, 48:58-62.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
1102

Matinkhoo, K., A. Pryyma, A.A.W.L. Wong, and D. Perrin.
(2021) Synthesis and evaluation of “ama-flash”, a
photocaged amatoxin prodrug for light-activated rna
pol ii inhibition and cell death. Chemical
Communications.
Moldenhauer, G., A.V. Salnikov, S. Lüttgau, I. Herr, J.
Anderl, and H. Faulstich. (2012) Therapeutic potential
of amanitin-conjugated anti-epithelial cell adhesion
molecule monoclonal antibody against pancreatic
carcinoma. JNCI: Journal of the National Cancer
Institute, 104:622-634.
Morgan, M.J. and Z.-g. Liu. (2011) Crosstalk of reactive
oxygen species and nf-κb signaling. Cell Research,
21:103-115.
Moshnikova, A., V. Moshnikova, O.A. Andreev, and Y.K.
Reshetnyak. (2013) Antiproliferative effect of phlip-
amanitin. Biochemistry, 52:1171-1178.
Nasiri, H., Z. Valedkarimi, L. Aghebati-Maleki, and J.
Majidi. (2018) Antibody-drug conjugates: Promising
and efficient tools for targeted cancer therapy. J Cell
Physiol, 233:6441-6457.
Osborne, J.R., N.H. Akhtar, S. Vallabhajosula, A. Anand,
K. Deh, and S.T. Tagawa. (2013) Prostate-specific
membrane antigen-based imaging. Urologic oncology,
31:144-154.
Rodrigues, D.F., R. Pires das Neves, A.T.P. Carvalho, M.
Lourdes Bastos, V.M. Costa, and F. Carvalho. (2020)
In vitro mechanistic studies on α-amanitin and its
putative antidotes. Arch Toxicol, 94:2061-2078.
Rudd, M.D. and D.S. Luse. (1996) Amanitin greatly
reduces the rate of transcription by rna polymerase ii
ternary complexes but fails to inhibit some transcript
cleavage modes. J Biol Chem, 271:21549-58.
Saravanapriya, P. and K.P. Devi, (year) Chapter 14 - plant
extracts with putative hepatotoxicity activity, in
Influence of nutrients, bioactive compounds, and plant
extracts in liver diseases, S.M. Alavian, et al., Editors.
2021, Academic Press. p. 259-287.
Shuptrine, C.W., R. Surana, and L.M. Weiner. (2012)
Monoclonal antibodies for the treatment of cancer.
Semin Cancer Biol, 22:3-13.
Steurer, B., R.C. Janssens, B. Geverts, M.E. Geijer, F.
Wienholz, A.F. Theil, J. Chang, S. Dealy, J. Pothof,
W.A. van Cappellen, A.B. Houtsmuller, et al. (2018)
Live-cell analysis of endogenous gfp-rpb1 uncovers
rapid turnover of initiating and promoter-paused rna
polymerase ii. Proceedings of the National Academy of
Sciences, 115:E4368-E4376.
Strassz, A., M.S. Raab, R.Z. Orlowski, M. Kulke, G.
Schiedner, and A. Pahl. (2020) A first in human study
planned to evaluate hdp-101, an anti-bcma amanitin
antibody-drug conjugate with a new payload and a new
mode of action, in multiple myeloma. Blood, 136:34.
Sung, H., J. Ferlay, R.L. Siegel, M. Laversanne, I.
Soerjomataram, A. Jemal, and F. Bray. (2021) Global
cancer statistics 2020: Globocan estimates of incidence
and mortality worldwide for 36 cancers in 185
countries. CA: A Cancer Journal for Clinicians, 71:209-
249.
Świderska, K.W., A. Szlachcic, Ł. Opaliński, M.
Zakrzewska, and J. Otlewski. (2018) Fgf2 dual
warhead conjugate with monomethyl auristatin e and α-
amanitin displays a cytotoxic effect towards cancer
cells overproducing fgf receptor 1. International
Journal of Molecular Sciences, 19:2098.
Van der Jeught, K., H.-C. Xu, Y.-J. Li, X.-B. Lu, and G. Ji.
(2018) Drug resistance and new therapies in colorectal
cancer. World journal of gastroenterology, 24:3834-
3848.
Wang, M., Y. Chen, Z. Guo, C. Yang, J. Qi, Y. Fu, Z. Chen,
P. Chen, and Y. Wang. (2018) Changes in the
mitochondrial proteome in human hepatocytes in
response to alpha-amanitin hepatotoxicity. Toxicon,
156:34-40.
Wang, Y.-M.C., B. Sloey, T. Wong, P. Khandelwal, R.
Melara, and Y.-N. Sun. (2011) Investigation of the
pharmacokinetics of romiplostim in rodents with a
focus on the clearance mechanism. Pharmaceutical
Research, 28:1931-1938.
WIELAND, T. (1983) The toxic peptides from amanita
mushrooms. International Journal of Peptide and
Protein Research, 22:257-276.
Wu, B., J. Johnson, M. Soto, M. Ponce, D. Calamba, and
Y.-N. Sun. (2012) Investigation of the mechanism of
clearance of amg 386, a selective angiopoietin-1/2
neutralizing peptibody, in splenectomized,
nephrectomized, and fcrn knockout rodent models.
Pharmaceutical Research, 29:1057-1065.
ZANOTTI, G., C. MÖHRINGER, and T. WIELAND.
(1987) Synthesis of analogues of amaninamide, an
amatoxin from the white amanita virosamushroom.
International Journal of Peptide and Protein Research,
30:450-459.
Zhao, L., J.P. May, A. Blanc, D.J. Dietrich, A. Loonchanta,
K. Matinkhoo, A. Pryyma, and D.M. Perrin. (2015)
Synthesis of a cytotoxic amanitin for biorthogonal
conjugation. ChemBioChem, 16:1420-1425.
Zheleva, A. (2013) Phenoxyl radicals formation might
contribute to severe toxicity of mushrooom toxin alpha
amanitin-an electron paramagnetic resonance study.
Trakia Journal of Sciences, 11:33-38.
Zheleva, A., A. Tolekova, M. Zhelev, V. Uzunova, M.
Platikanova, and V. Gadzheva. (2007) Free radical
reactions might contribute to severe alpha amanitin
hepatotoxicity – a hypothesis. Medical Hypotheses,
69:361-367.
Toxic Mechanisms of -Amanitin and Its Potential to Fight Cancer
1103
