
Hypotheses of Alzheimer’s Disease Pathogenesis
Liuhua Chen
Departments of Botany and Zoology, University of British Columbia, Vancouver, V6T 1Z1, Canada
Keywords:
Alzheimer’s Disease, Amyloid Aggregation, Tau, APP, Senile Plaque, NFT.
Abstract: Alzheimer’s disease (AD) is the most common cause of dementia. The two hallmarks of AD are extracellular
senile plaques (SF) and intracellular neurofibrillary tangles (NFT). Numerous studies have been involved in
research for AD pathogenesis, proposing different hypotheses and theories to explain the occurrence of the
two hallmarks as well as AD onset. This paper will review three suppositions of AD pathogenesis, including
the amyloid cascade hypothesis, the APP metabolism impairment theory, and Tau hypothesis. The amyloid
cascade hypothesis is the mainstream hypothesis that suggests Aβ aggregation should predate all AD-related
pathological events. Amyloid precursor protein (APP) metabolism theory considers impaired metabolism of
APP to be a possible explanation for AD onset. Tau hypothesis, which is another major hypothesis for AD
pathogenesis, postulates that tau aggregation and NFT play an initiating role in AD onset. Many studies have
elucidated that Aβ aggregation may not cause AD; some of the AD researchers have shifted their research
focus to alternative hypotheses involving other biomarkers, such as tau and APP metabolites. However, the
role of Aβ should not be refuted as it interacts with so many AD pathological features. Research on Aβ should
be alongside explorations of other potential pathological causes of AD.
1 INTRODUCTION
Alzheimer’s disease (AD) is a progressive and
irreversible neurodegenerative disease that is
affecting millions of people in the world. AD is
categorized into two divisions based on the onset age.
Sporadic AD or late-onset AD is the most common
type of AD; it usually manifests after age 65. Familial
AD, also known as early-onset AD, occurs in people
younger than age 60. Both types of AD together
account for more than half of the dementia which is
recognized as a public health priority by WHO. In the
past, most of the early-staged dementia used to be
deemed as one of the normal consequences of aging,
overlooking AD onset. However, AD is not an
inevitable consequence of aging but an abnormal
neurological disorder that will cause more severe
cognitive impairment as the disease progresses.
Two hallmarks of AD are extracellular senile
plaques (SP) and intracellular neurofibrillary tangles
(NFT) within the central nervous system. There are
numerous studies on SP, NFT, and the relevant
metabolic processes to investigate AD pathologies;
however, different hypotheses have not yet unified to
provide a clear AD pathology because of the complex
nature of AD and the limitations of the hypotheses. It
is of importance to summarize and compare current
studies on AD pathogenesis in order to unravel the
full picture of AD mechanism. This paper reviews
several hypotheses of AD pathogenesis, including the
amyloid cascade hypothesis, APP metabolism theory,
and tau hypothesis, to serve as a concise summary of
the current progress in AD mechanism research.
2 AMYLOID CASCADE
HYPOTHESIS
The amyloid cascade hypothesis, also known as beta-
amyloid hypothesis, posits that SPs are amyloid
plaques, which are mainly composed of Aβ fibrils,
contribute to NFT and other neuronal alternations
associated with AD-induced dementia (Jack, Jr., et al.
2016, Bondi, Edmonds, and Salmon 2017) . For a
long time, Aβ aggregation was positioned as the
upstream cause of all the pathological changes in AD.
Aβ is a metabolic product of amyloid-beta precursor
protein (APP) which plays important role in neuronal
development, neurite outgrowth, as well as
intracellular trafficking in axons (Kametani, and
Hasegawa 2018). APP (Figure 1) has four cleavage
478
Chen, L.
Hypotheses of Alzheimer’s Disease Pathogenesis.
DOI: 10.5220/0011372500003438
In Proceedings of the 1st International Conference on Health Big Data and Intelligent Healthcare (ICHIH 2022), pages 478-483
ISBN: 978-989-758-596-8
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
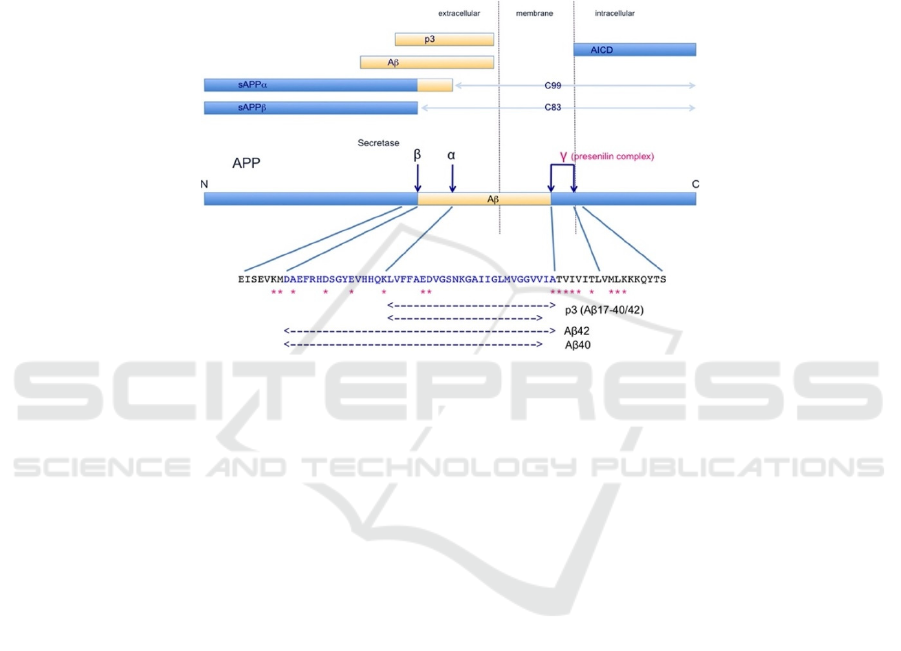
sites (i.e. α, β, and two γ sites) respectively
recognized and cleaved by α-secretase, β-secretase,
and γ secretase. Aβ is produced by the cleavages of
APP by β-secretase and γ secretase. In accordance
with the two γ cleavage sites on APP, β-γ
combinatorial cleavage produces either Aβ40 or
Aβ42, depending on which γ site the secretase binds.
The research suggests that the elevated production
level of Aβ42 rather than Aβ40 leads to aggregation
of SP (Hillen 2019). Normally, Aβ is released out of
the neurons and degraded; however, in pathological
conditions, Aβ is accumulated to form extracellular
plaques. Moreover, Aβ exists in three forms, which
are soluble Aβ monomers, soluble Aβ oligomer, and
insoluble Aβ fibrils. The amyloid cascade hypothesis
considers amyloid plaques the aggregation of the
insoluble Aβ fibrils.
Figure 1. APP metabolism and the metabolic products. Four dark blue arrows are secretase recognition sites on APP; α, β,
and γ sites correspond to α-, β-, and γ-secretases respectively. P3 is produced by cleavage by α and γ secretase. Aβ is produced
by β and γ secretase. sAPPα and sAPPβ are produced by α-secretase and β-secretase cleavage respectively. AICD (APP
intracellular Domain) are C-terminal fragments that result from cleavage of γ secretase. Adapted from Kametani, F., &
Hasegawa, M. (2018). Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer's Disease. Front Neurosci,
12, 25. https://doi.org/10.3389/fnins.2018.00025.
The Amyloid cascade hypothesis initially
proposes a clear temporal order in the AD
pathogenesis, in which high-level aggregation of Aβ
is the causative agent of other pathological events
including NFT occurrence (Bondi, Edmonds, and
Salmon 2017, Hillen 2019). Cytotoxicity of Aβ
aggregation can directly cause neurodegeneration and
neuron loss (Kametani, and Hasegawa, Ohshima, et
al. 2018). Also, amyloid plaques can activate
microglia and astrocytes to release inflammatory
factors, causing neuron inflammation (Zhang, and
Zheng 2019). The activated microglia are phagocytic
towards synapses, resulting in synaptic impairment.
Combining the clinical examples that relate AD onset
to Aβ aggregation, β amyloid hypothesis is long
believed to primarily account for AD onset. However,
along with AD research advancing, amyloid cascade
hypothesis is challenged and disapproved by many
recent studies.
A lot of research, ranging from investigating Aβ
neurotoxicity to exploring the relationship between
AD and amyloid plaques, has validated that the
temporal ordering proposed in the amyloid cascade
hypothesis is wrong ((Kametani, and Hasegawa 2018,
Hillen 2019). Jack et al. have acknowledged that
temporal ordering of amyloid cascade hypothesis is
problematic. In terms of Aβ toxicity, several lines of
evidence have rejected that Aβ cytotoxicity causes
synapse loss or neurodegeneration (Bondi, Edmonds,
and Salmon 2017). Braak et al. directly refute the
temporal ordering of amyloid cascade hypothesis by
reporting that NFT is not a downstream event led by
amyloid plaques, and that tau aggregation,
components of NFT, precedes formation of amyloid
plaques. In addition, amyloid plaques are not always
related to AD as normal people could have more
extensive amyloid aggregation and same-aged AD
patients and normal people could have amyloid
plaques of the same density (Edison, et al. 2007). It
comes to a conclusion that amyloid plaque or senile
plaque aggregation is related to aging but may be
irrelevant to AD onset. Furthermore, the most
convincing evidence refusing amyloid cascade
hypothesis might be the devastating failure of all
Hypotheses of Alzheimer’s Disease Pathogenesis
479

clinical trials on AD therapeutics aiming to stop or
delay AD progress by eliminating or preventing Aβ
aggregation. β-secretase inhibitors that block the
cleavage of Aβ from APP should have reduced Aβ
production and aggregation; on the contrary, groups
treated with the β-secretase inhibitors show more
severe cognitive impairment than the control
(Knopman 2019). In conclusion, it is unlikely that
amyloid cascade hypothesis is a correct AD
pathogenic mechanism. Yet rejecting the amyloid
cascade hypothesis should not discontinue the
research on Aβ, Aβ aggregation might still be one of
the pathological events of AD onset.
3 APP METABOLISM
IMPAIRMENT THEORY
Amyloid-beta precursor protein (APP) metabolism,
also known as APP processing, generates several
downstream peptides that are found to be important
for neuron functionality. Some research suggest that
AD pathogenesis should be related to the impairment
of APP metabolism, where all the major APP
metabolites should be considered beside Aβ
aggregation (Bondi, Edmonds, and Salmon 2017,
Kametani, and Hasegawa 2018). APP gene locates on
chromosome 21 of which the trisomy form causes the
famous Down’s Syndrome. Patients of Down’s
Syndrome also exhibit AD-like symptoms such as
cognitive ability impairment, which could be caused
by the excess copies of APP gene and high amount of
APP metabolites including Aβ (Wilson, et al. 2019).
APP can be processed by three different
secretases (i.e. α-secretase, β-secretase, and γ
secretase) via four recognition sites (i.e. α, β, and two
γ sites) (Figure 1). In the non-amyloidogenic
pathway, APP is sequentially cleaved by α-secretase
and γ-secretase; in the amyloidogenic pathway, α-
secretase is substituted by β-secretase, generating Aβ.
In both pathways, γ-secretase cleaves to produce APP
intracellular domain (AICD). Evidence suggests that
α-secretase predominately cleaves more than 90% of
the APP while β-secretase only accounts for less than
10% of the APP cleavage (Kametani, and Hasegawa
2018). Accordingly, the major metabolites should be
sAPPα, p3, C99, and AICD while Aβ is supposed to
be in a small amount (Figure 1). In pathological
conditions, a large amount of Aβ is produced to form
amyloid aggregation, indicating that β cleavage is
more predominant than α cleavage. There is another
explanation for the prevalence of β cleavage.
Research on APP trafficking found that APP
primarily locates intracellularly, co-residing with β-
secretase whose activity is optimized by the acidic
endosomal environment, causing amyloidogenic
processing (Wang, et al. 2017). On the contrary, only
a small fraction of APP, localizing on the cell surface
where α-secretase is abundant, undergoes non-
amyloidogenic pathway. Given that β-secretase is
pivotal to Aβ generation, inhibiting β cleavage should
have ameliorated AD symptoms by reducing Aβ
production. However, experiment of β cleavage
inhibition, which aims to reduce Aβ aggregation,
even worsens cognitive impairment (Knopman
2019), implying that AD pathology is not limited to
amyloid plaques but APP metabolism as a whole.
Besides the notorious Aβ accumulation in the
amyloidogenic pathway, research has also
investigated the neuronal effects of other APP
metabolites (i.e. sAPPα, p3, C99, and AICD) of the
non-amyloidogenic pathway (Zhang, et al. 2011).
sAPPα
is considered to be neurotrophic (Zhang, and
Zheng 2019). P3 has long been classified as “non-
amyloidogenic”, so studies on APP metabolites
usually omit p3 and few studies focus on its
cytotoxicity. Nevertheless, Kuhn et al. and Kuhn and
Raskatov have revisited the function of p3; they
concluded that p3 is amyloidogenic and it may play a
role in amyloid plaque formation. Firstly, p3 contains
the amyloidogenic region in Aβ; secondly, p3 fibrils
facilitate the formation of Aβ fibrils that later
assemble to become amyloid plaques (Kuhn and
Raskatov 2020). Some researchers believe that Aβ
toxicity is due to the hydrophobicity of Aβ oligomers
(Hardy, and Selkoe 2002). P3 is almost entirely
hydrophobic, so it could have higher cytotoxicity than
Aβ (Wei, et al. 2002), which might eventually
contribute to neurodegeneration.
C99 and AICD are both C-terminal fragments
(CTF) of APP. CFT accumulation might be closely
related to AD onset. Stocking of CTF could induce
synaptic failure, abnormally phosphorylated tau
protein, and memory loss (Tamayev, et al. 2012) .
Additionally, the accumulation of CFT interferes the
normal function and morphology of mitochondria in
neurons (Vaillant-Beuchot, et al. 2021, Devi, et al.,
2006); specifically, CFT accumulation alters
mitochondria sizes, disorganize mitochondrial
cristae, and affects the mitophagy process.
Mitochondria are believed to be involved in AD
pathologies. Nevertheless, its role in the pathogenesis
is unclear. Some research proposes a primary
mitochondria hypothesis where mitochondria
disruption causing neuron dysfunction predates Aβ
accumulation. Meanwhile, some others studies
support a secondary mitochondria hypothesis in
ICHIH 2022 - International Conference on Health Big Data and Intelligent Healthcare
480
which mitochondrial dysfunction is downstream of
Aβ aggregation (Devi, et al. 2006, Swerdlow 2018).
Summing the studies on APP metabolites, it is
clear that Aβ is not the only neurotoxic APP
metabolite that could potentially lead to
neurodegeneration. The impairment of APP
processing that alters the relative amount of various
APP metabolites should be considered as a whole to
explain AD onset.
4 TAU HYPOTHESIS
Tau is a microtubule-associated protein that is
responsible for the regulation of tubulin assemblies
and stabilization of neuronal microtubules in the
central nervous system. Normally, tau is only
moderately phosphorylated; hyperphosphorylated tau
fibrils form NFT, which is an AD hallmark. Tau’s
hypothesis suggests that tau in hyperphosphorylated
states form paired helical filaments and straight
filaments, both of which contribute to the formation
of intracellular NFT ((Kametani, and Hasegawa
2018, Muralidar, et al. 2020) . In tau hypothesis, Aβ
is a tau-induced downstream event.
Tau is cytotoxic as it negatively affects neuron
cytoskeletal structure, axonal transport, and
mitochondrial membrane integrity.
Hyperphosphorylated tau loses the ability to regulate
tubulin, which is pivotal for the neuron skeletal
system (Muralidar, et al. 2020). One of the most
important functions of tau is stabilizing the
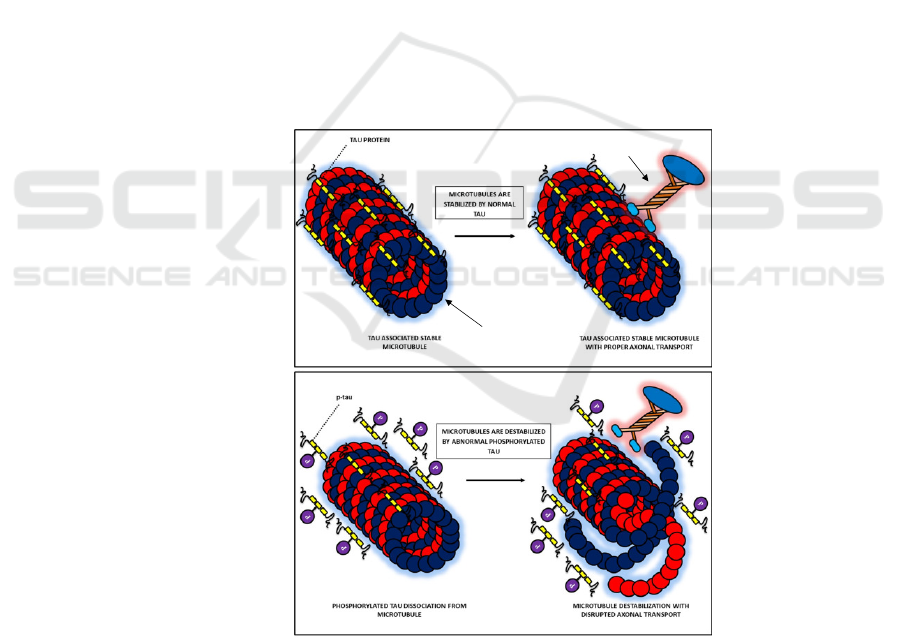
microtubules in the axons. Normally, moderate
phosphorylated tau attaches to the microtubules in the
axons, allowing for the binding of motor molecules
that act as cargo for intracellular trafficking (Combs,
et al. 2019) (Figure 2a). Hyperphosphorylated tau
detaches from the microtubules (Figure 2b),
disaggregating the microtubules and thus disrupting
the axonal transport (Combs, et al. 2019, Arnsten, et
al. 2021); later, the detached tau assembles into NFT.
Disruption of axonal transport eventually contribute
to neurodegeneration. (Camilleri, Ghio) report that
accumulation of tau leads to mitochondrial organelle
swelling and loss of membrane potential, causing
mitochondrial defects
Figure 2. Stabilization of microtubules by tau protein. (a) In normal neurons, moderately phosphorylated tau (without P)
proteins associate to stabilize the microtubules, allowing for the binding of motor molecules. (b). Hyperphosphorylated tau
(with P) detach from microtubules, which then disaggregate microtubular structure. Motor molecules fail to bind to the
microtubules. Adapted from Muralidar, S., Ambi, S. V., Sekaran, S., Thirumalai, D., & Palaniappan, B. (2020). Role of tau
protein in Alzheimer's disease: The prime pathological player. Int J Biol Macromol, 163, 1599-1617.
a
b
Microtubules
Motor Molecules
Hypotheses of Alzheimer’s Disease Pathogenesis
481

The core proposition of tau hypothesis is that the
tau is the causative agent of AD and it is upstream of
Aβ plaques. Multiple studies have confirmed the
upstream tole of tau and verified tau-induced NFT
(Arnsten, et al. 2021) . Most importantly, the self-
propagating and aggregation-promoting
characteristics of tau can explain the staging and
progression of AD. Abnormally hyperphosphorylated
tau occurs in only a limited range of neurons, and they
can convert the normal tau into the
hyperphosphorylated states, increasing the total
amount of hyperphosphorylated tau and propagating
to a larger range of neurons (Nonaka, et al. 2010).
Moreover, aggregation of hyperphosphorylated tau
inhibits protein aggregation clearance, which in turn
protects tau aggregation from degradation (Keller,
Hanni, and Markesbery 2000), forming a viscous
cycle that enables for enlargement of
hyperphosphorylated tau aggregation. The nature of
the viscous cycle and self-propagation of tau might
explain the progression of AD clinically.
5 CONCLUSIONS
In summary, different hypotheses have not unified to
provide a clear AD pathology yet. A large amount of
evidence has rejected the amyloid cascade
hypothesis, especially the temporal ordering.
However, the role of Aβ should not be completely
refuted since it interacts with so many other
hypotheses for AD pathology. Some other
hypotheses, such as APP metabolism theory and Tau
hypothesis, seem to be valid explanations for AD. P3
and CTF, which are under-researched APP
metabolites, should be revisited for their
neurotoxicity, their relationship to SF and NFT, and
the relevance to AD onset. Also, as evidence
suggests, tau aggregation should be one of the pivotal
events in AD pathology that needs further
investigation. This review collates the above
hypotheses of AD pathogenesis and provides a clear
demonstration and comparison of the current
advances in AD research, which provide a wide
picture for AD mechanisms. To date, we have
witnessed an explosion of research into Alzheimer's
disease and the development of drugs at all levels, but
much remains to be done. As mentioned above, the
failure of clinical trials suggests that we should revisit
the role of beta-amyloid and reconsider other factors
involved in AD pathogenesis in future research.
ACKNOWLEDGMENTS
I would like to thank Jiaqiong Sun for providing
instructions on developing paper outlines and Min
Han for providing advice on editing. I would want to
express my gratitude to Dr. Kate Jeffery for offering
suggestions on literature research on Alzheimer’s
disease. Lastly, I would also like to thank my parents
and all of my friends for all the encouragement and
support all the time.
REFERENCES
Arnsten, A.F.T., et al., Hypothesis: Tau pathology is an
initiating factor in sporadic Alzheimer's disease.
Alzheimers Dement, 2021. 17(1): p. 115-124.
Bondi, M.W., E.C. Edmonds, and D.P. Salmon,
Alzheimer's Disease: Past, Present, and Future. J Int
Neuropsychol Soc, 2017. 23(9-10): p. 818-831.
Braak, H., et al., Stages of the pathologic process in
Alzheimer disease: age categories from 1 to 100 years.
J Neuropathol Exp Neurol, 2011. 70(11): p. 960-9.
Camilleri, A., et al., Tau-induced mitochondrial membrane
perturbation is dependent upon cardiolipin. Biochim
Biophys Acta Biomembr, 2020. 1862(2): p. 183064.
Combs, B., et al., Tau and Axonal Transport Misregulation
in Tauopathies. Adv Exp Med Biol, 2019. 1184: p. 81-
95.
Devi, L., et al., Accumulation of amyloid precursor protein
in the mitochondrial import channels of human
Alzheimer's disease brain is associated with
mitochondrial dysfunction. J Neurosci, 2006. 26(35): p.
9057-68.
Edison, P., et al., Amyloid, hypometabolism, and cognition
in Alzheimer disease: an [11C]PIB and [18F]FDG PET
study. Neurology, 2007. 68(7): p. 501-8.
Hardy, J. and D.J. Selkoe, The amyloid hypothesis of
Alzheimer's disease: progress and problems on the road
to therapeutics. Science, 2002. 297(5580): p. 353-6.
Hillen, H., The Beta Amyloid Dysfunction (BAD)
Hypothesis for Alzheimer's Disease. Front Neurosci,
2019. 13: p. 1154.
Jack, C.R., Jr., et al., A/T/N: An unbiased descriptive
classification scheme for Alzheimer disease
biomarkers. Neurology, 2016. 87(5): p. 539-47.
Kametani, F. and M. Hasegawa, Reconsideration of
Amyloid Hypothesis and Tau Hypothesis in
Alzheimer's Disease. Front Neurosci, 2018. 12: p. 25.
Keller, J.N., K.B. Hanni, and W.R. Markesbery, Impaired
proteasome function in Alzheimer's disease. J
Neurochem, 2000. 75(1): p. 436-9.
Knopman, D.S., Lowering of Amyloid-Beta by beta-
Secretase Inhibitors - Some Informative Failures. N
Engl J Med, 2019. 380(15): p. 1476-1478.
Kuhn, A.J. and J. Raskatov, Is the p3 Peptide (Abeta17-40,
Abeta17-42) Relevant to the Pathology of Alzheimer's
Disease?1. J Alzheimers Dis, 2020. 74(1): p. 43-53.
ICHIH 2022 - International Conference on Health Big Data and Intelligent Healthcare
482

Kuhn, A.J., et al., Alzheimer's Disease "Non-
amyloidogenic" p3 Peptide Revisited: A Case for
Amyloid-alpha. ACS Chem Neurosci, 2020. 11(11): p.
1539-1544.
Muralidar, S., et al., Role of tau protein in Alzheimer's
disease: The prime pathological player. Int J Biol
Macromol, 2020. 163: p. 1599-1617.
Nonaka, T., et al., Seeded aggregation and toxicity of
{alpha}-synuclein and tau: cellular models of
neurodegenerative diseases. J Biol Chem, 2010.
285(45): p. 34885-98.
Ohshima, Y., et al., Mutations in the beta-amyloid
precursor protein in familial Alzheimer's disease
increase Abeta oligomer production in cellular models.
Heliyon, 2018. 4(1): p. e00511.
Swerdlow, R.H., Mitochondria and Mitochondrial
Cascades in Alzheimer's Disease. J Alzheimers Dis,
2018. 62(3): p. 1403-1416.
Tamayev, R., et al., beta- but not gamma-secretase
proteolysis of APP causes synaptic and memory deficits
in a mouse model of dementia. EMBO Mol Med, 2012.
4(3): p. 171-9.
Vaillant-Beuchot, L., et al., Accumulation of amyloid
precursor protein C-terminal fragments triggers
mitochondrial structure, function, and mitophagy
defects in Alzheimer's disease models and human
brains. Acta Neuropathol, 2021. 141(1): p. 39-65.
Wang, X., et al., Modifications and Trafficking of APP in
the Pathogenesis of Alzheimer's Disease. Front Mol
Neurosci, 2017. 10: p. 294.
Wei, W., et al., Abeta 17-42 in Alzheimer's disease
activates JNK and caspase-8 leading to neuronal
apoptosis. Brain, 2002. 125(Pt 9): p. 2036-43.
Wilson, L.R., et al., Differential effects of Down's
syndrome and Alzheimer's neuropathology on default
mode connectivity. Hum Brain Mapp, 2019. 40(15): p.
4551-4563.
Zhang, H. and Y. Zheng, [beta Amyloid Hypothesis in
Alzheimer's Disease: Pathogenesis, Prevention, and
Management]. Zhongguo Yi Xue Ke Xue Yuan Xue
Bao, 2019. 41(5): p. 702-708.
Zhang, Y.W., et al., APP processing in Alzheimer's disease.
Mol Brain, 2011. 4: p. 3.
Hypotheses of Alzheimer’s Disease Pathogenesis
483
