
Mechanisms and Electron Transfer of Prokaryotic Nitrate
Reductases
Yiming Luo
Hwa Chong International School, Singapore, 269783, Singapore
Keywords: Prokaryotic Nitrate Reductases, Metalloenzyme, Special Cofactors, Electron Transfer.
Abstract: There are three types of prokaryotic nitrate reductases which are essential in the biological nitrogen cycle,
respectively membrane-bound respiratory nitrate reductase (Nar), periplasmic nitrate reductase (Nap) and
prokaryotic assimilatory nitrate reductase (Nas). This work summarizes the structures of these three types of
nitrate reductases, as well as their nitrate reduction mechanism and electron transfer pathways. They each
contain a molybdenum bis-molybdopterin guanine dinucleotide cofactor (Mo-bis-MGD) as the active site for
nitrate binding, and they use different numbers and types of iron–sulfur clusters to aid the electron transfer.
Nar and Nap use hemes and the quinol–quinone Q cycle as the electron source, whereas Nas relies on
flavodoxin or NADH. This work also presents some possible aspects for future research, such as the
uniqueness of molybdenum, the large potential barriers in the Nap electron transfer pathway, and unsolved
crystal structures of some enzyme subunits that have limited understanding.
1 INTRODUCTION
Nitrogen is an element necessary for all lifeforms
because it is a building block of many essential
biological molecules such as nucleic acids, amino
acids, and proteins. It occurs in various oxidation
states, such as dinitrogen (N
2
, 0) in the atmosphere,
ammonium (NH
4
+
, −3) in sediments and minerals,
nitrate (NO
3
−
, +5) as dissolved species in marine
environments (Bebout, Fogel, Cartigny, 2013), and
organic nitrogen compounds in organisms. Nitrogen
can be converted between these forms, referred to as
the nitrogen cycle. Nitrate reductases play an
essential role in the nitrogen cycle, by catalyzing the
reduction of nitrate to nitrite (NO
2
−
), which is ready
for further reduction:
NO
3
−
+2H
+
+ 2e
−
→ NO
2
−
+ H
2
O, E
⦵
= +
420
mV
(Equation 1)
The aim of this report is to provide a brief
introduction of the nitrogen cycle and nitrate
reduction pathways, summarize and compare the
structures of different prokaryotic nitrate reductases
and explore the mechanisms of the Mo-bis-MGD
active site, as well as the role of each special cofactor
in the entire electron transport chain.
2 COMMON METHODS TO
DETERMINE NITRATE
REDUCTASE STRUCTURE
Nitrate reductases are a category of metalloenzyme
with a molybdenum ion at each of their active sites.
Common methods to determine the structure of
nitrate reductases include X-ray crystallography,
specifically multiwavelength anomalous diffraction
(MAD), which provided high-resolution electron
density maps for the first crystal structure of
periplasmic nitrate reductase (Nap) (Dias, 1999).
This identified some unique cofactors present in
nitrate reductases: a molybdenum bis-molybdopterin
guanine dinucleotide cofactor (Mo-bis-MGD), and
an iron–sulfur cluster [4Fe–4S] (Figure 1). To
understand the coordination and redox activities of
the central Mo ion, electron paramagnetic resonance
(EPR) spectroscopy and extended X-ray absorption
fine structure (EXAFS) spectroscopy were used,
which facilitated the investigation of mechanisms of
catalysis (Butler, 1999).
Luo, Y.
Mechanisms and Electron Transfer of Prokaryotic Nitrate Reductases.
DOI: 10.5220/0011219400003443
In Proceedings of the 4th International Conference on Biomedical Engineering and Bioinformatics (ICBEB 2022), pages 505-513
ISBN: 978-989-758-595-1
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
505
3 THE BIOLOGICAL NITROGEN
CYCLE AND NITRATE
REDUCTASES
Nitrogen can be converted between different forms
through abiotic processes such as the production of
nitrogen monoxide (NO) by lightning:
N
2
+ O
2
→ 2NO (Equation 2)
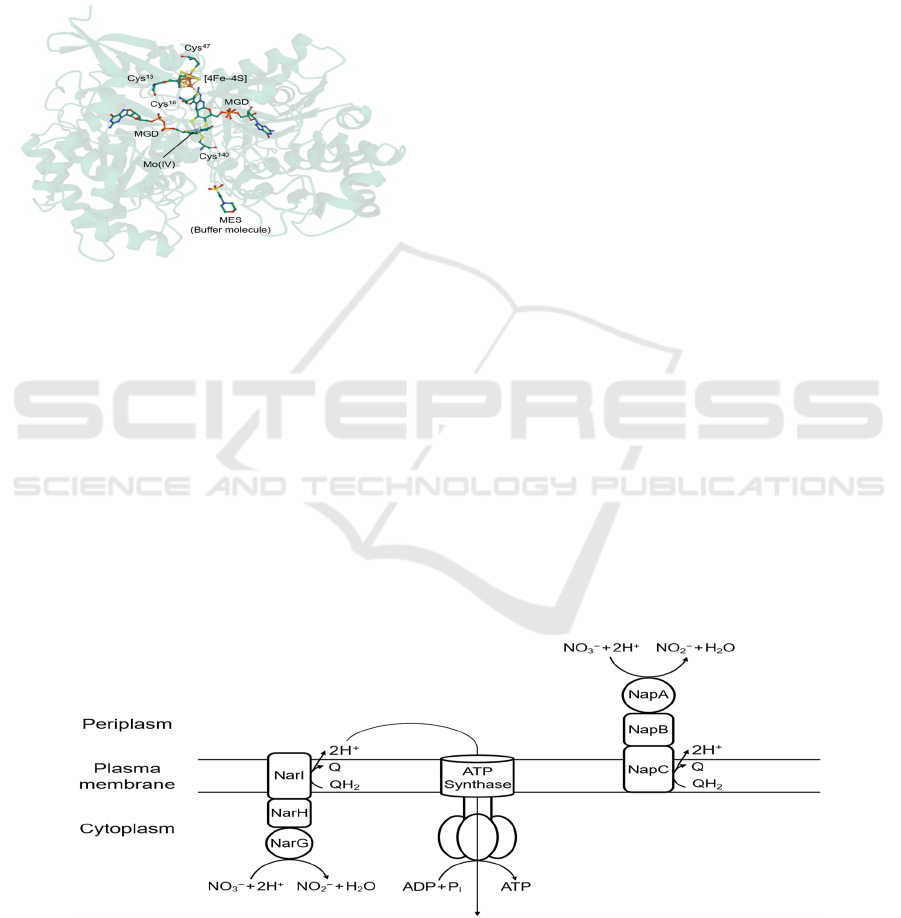
Figure 1: First crystal structure of Nap (subunit NapA) of
bacterium Desulfovibrio desulfuricans (PDB: 2NAP)
obtained by MAD methods. The central Mo (IV) ion is
coordinated to two MGDs to form a Mo-bis-MGD cofactor,
as well as a cysteine residue Cys
140
. An iron–sulfur cluster
[4Fe–4S] is located nearby for electron transfer.
However, the focus of this report will be the
biological nitrogen cycle, where nitrogen is
circulated in the biosphere by organisms,
predominantly prokaryotes. Different nitrogen
compounds are simultaneously oxidized or reduced,
and the reductive process can be further divided into
two different pathways: nitrate assimilation and
denitrification (Cabello, 2009), or dissimilatory
nitrate reduction. In nitrate assimilation, NO
3
−
are
reduced to NH
4
+
, a nitrogen source for constructive
metabolism, via the only intermediate of NO
2
−
,
catalyzed by assimilatory nitrate reductase (Nas) and
nitrite reductase NirS:
NO
3
−
Nas
⎯→⎯⎯
NO
2
−
NirS
⎯→⎯⎯
NH
4
+
(Equation 3)
In contrast to nitrate assimilation, denitrification
refers to the catabolic process where NO
3
−
acts as the
terminal electron acceptor in anaerobic respiration
instead of dioxygen (O
2
). The terminal products of
denitrification can be either nitrous oxide (N
2
O) or
N
2
, via the intermediate stages of NO
2
−
and NO. In
this case, NO
3
−
is reduced to NO
2
−
with the catalysis
of either Nap or membrane-bound respiratory nitrate
reductase (Nar), and the subsequent reduction are
catalyzed respectively by nitrite reductases NirS and
NirK, NO reductases cNor and qNor, and N
2
O
reductase (Nos):
NO
3
−
Nar/ Nap
⎯→⎯⎯⎯
NO
2
−
NirS/ NirK
⎯→⎯⎯⎯
NO
cNor/qNor
⎯→⎯⎯⎯
N
2
O
Nos
⎯→⎯⎯
N
2
(Equation 4)
This report will only discuss prokaryotic nitrate
reductases, i.e. Nas, Nap and Nar, among these
enzymes in nitrate reduction processes. A key
difference between Nap and Nar is that the active site
Mo-bis-MGD of Nap is in the periplasmic space,
while Nar’s is in the cytoplasm (Moreno-Vivián,
1999). Nar consumes protons to reduce NO
3
−
in the
cytoplasm and protons are translocated into the
periplasm through the oxidation of quinols to
quinones (QH
2
→ Q + 2H
+
+ 2e
−
) in the lipid bilayer
by the quinol-oxidizing subunit NarI, thereby
enhancing the electrochemical proton gradient to
facilitate adenosine triphosphate (ATP) synthesis,
while Nap reduces NO
3
−
in the periplasm so there is
no proton translocation (Kuypers, 2018) (Figure 2).
Figure 2: Difference in location and contribution of ATP synthesis between Nar and Nap. The active sites where Mo-bis-
MGD is present are represented by circles, i.e. NarG and NapA.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
506
4 SPECIAL COFACTORS
PRESENT IN PROKARYOTIC
NITRATE REDUCTASES
4.1 Mo-Bis-MGD
Mo-containing enzymes can be categorized into 3
families with similar general structures, all of which
contain at least one MGD molecule each (Figure 3),
respectively the xanthine oxidase family, the
dimethyl sulfoxide (DMSO) reductase family, and
the sulfite oxidase family (Moura, 2004) (Figure 4a).
Though eukaryotic nitrate reductases (eukNR)
belong to the sulfite oxidase family, all prokaryotic
nitrate reductases are from the DMSO reductase
family, where the central Mo ion is coordinated to
two MGD molecules and one or two other ligands
(Sparacino-Watkins, 2014) (Figure 4b).
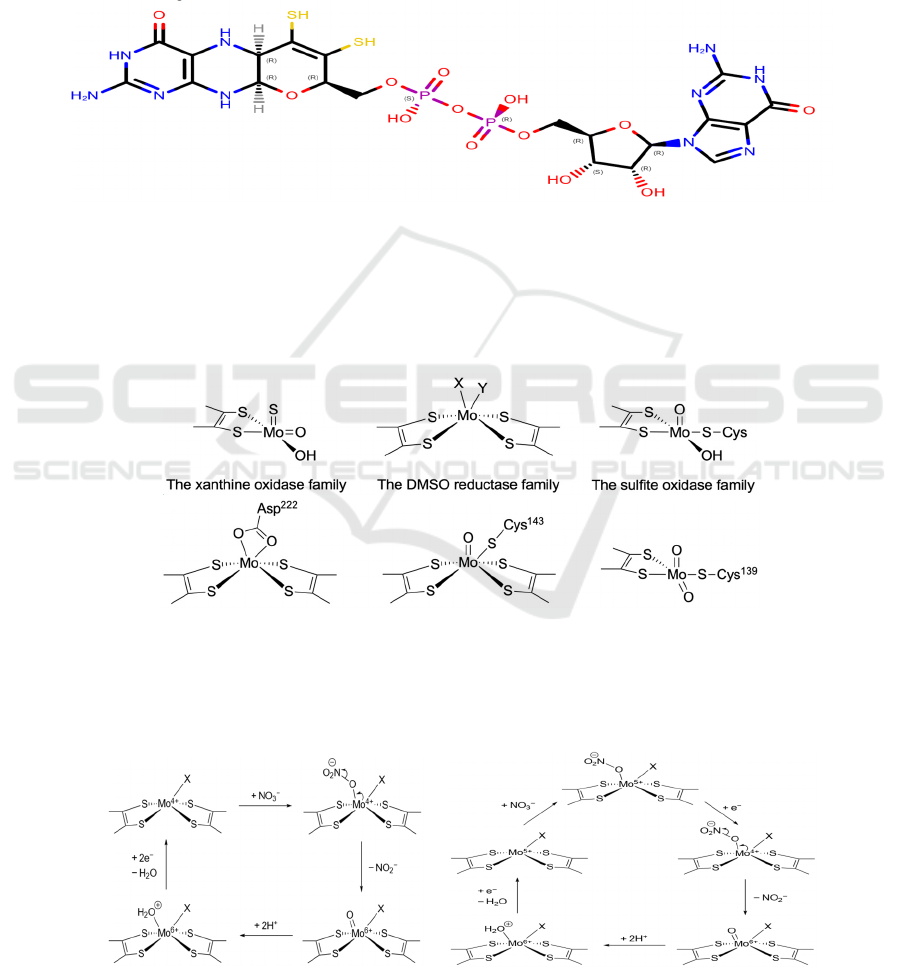
Figure 3: The structure of MGD.
The two sulfhydryl groups (colored yellow in
Figure 3) are responsible for Mo-coordination.
Although thiols generally have no net charge in
neutral pH, sulfhydryl groups are deprotonated and
coordinate as thiolates in Mo-bis-MGD (Moura,
2004). This is because highly charged Mo ions can
greatly stabilize the conjugate base of thiols, thus
lowering the pK
a
value of sulfhydryl groups in MGD
(Lippard, 1994).
4a
4b
Figure 4: (a) General structures of the three Mo-containing enzyme families. X and Y represent any ligand, either singly or
doubly bonded to the Mo ion. The −OH ligands may be replaced by −OH
2
or =O. (b) Some examples of Mo active sites.
From left to right: NarG (oxidized form) from Escherichia coli (PDB: 1Q16), NapA (oxidized form) from Escherichia coli
(PDB: 2NYA), eukNR from Pichia angusta (PDB: 2BII). The first two (Nap, Nar) belong to the DMSO reductase family
while the last one (eukNR) belongs to the sulfite oxidase family.
5a 5b
Figure 5: Two different proposed mechanisms of nitrate reduction at the Mo-bis-MGD active site. Though both mechanisms
have been proposed, Figure 5b shows the more widely accepted mechanism.
Mechanisms and Electron Transfer of Prokaryotic Nitrate Reductases
507
The central Mo ion has variable oxidation states
of IV, V and VI. Two different mechanisms of nitrate
reduction at the Mo-bis-MGD active site have been
proposed (Sparacino-Watkins, 2014) (Figure 5a and
5b). Nitrate reduction starts from the reduced form,
where the Mo ion is in the oxidation state of IV or V
and is only penta-coordinated with an empty binding
site for NO
3
−
. Mo(V) ion will then gain an electron,
because the binding of NO
3
−
raises its redox
potential, making it more easily reduced (Richardson,
2007) (Figure 5b). Since nitrogen is reduced from +5
to +3, the coordinated NO
3
−
gains two electrons from
Mo(IV), oxidizing it to be Mo(VI), with electron
configuration [Kr]. The Mo-bis-MGD cofactor is
now in its oxidized form, where an oxo ligand is left
coordinated to Mo(VI). It is then protonated and
leaves as water, reducing Mo(VI) back to either
Mo(IV) (Figure 5a) or Mo(V) (Figure 5b) for further
reduction of NO
3
−
. Regardless of the pathway, two
electrons in total are transferred in each cycle.
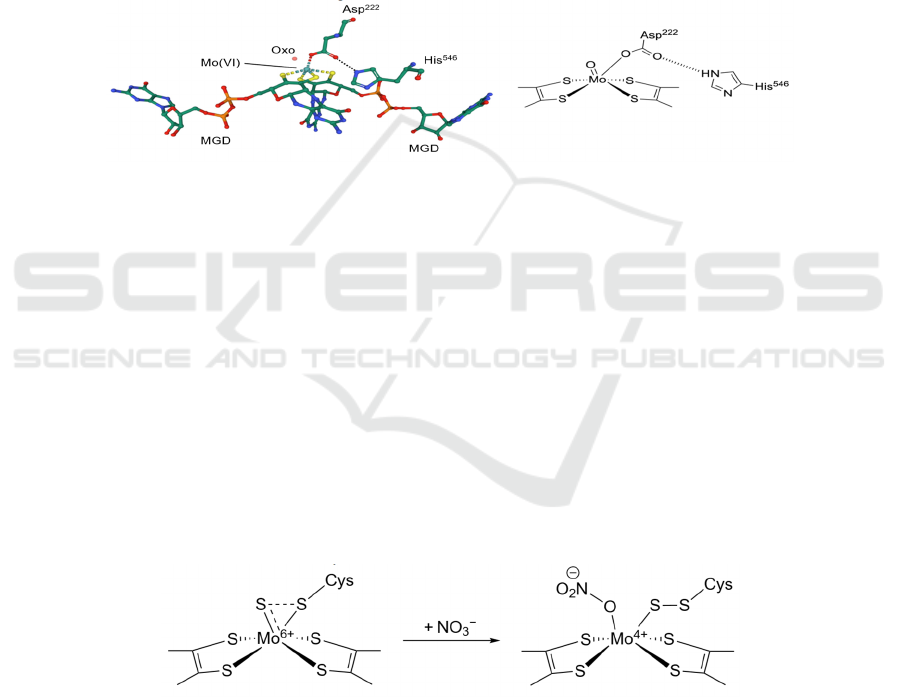
However, in the crystal structure of E. coli NarG
(PDB: 1Q16, leftmost in Figure 4b), a bidentate
Asp
222
was discovered, which might block the
binding of NO
3
−
. Besides, it was concluded that the
active site was in its oxidized form, i.e. Mo(VI), but
the oxo ligand that appeared in the proposed
mechanism was absent. In fact, there are different E.
coli NarG Mo-bis-MGD structures in the Protein
Data Bank (Figure 6).
Figure 6: A different structure of the Mo-bis-MGD active site of E. coli NarG (PDB: 1R27).
Figure 6 shows another oxidized form of the
active site, where the bidentate Asp
222
is now only
singly coordinated to the Mo ion, and the other
carboxylate O is hydrogen-bonded to the ε-nitrogen
of His
546
. Although the two structures are different,
their amino acid sequences are identical. The
difference may be caused by the protonation state of
His
546
, or structural flexibility of the active site.
However, further investigation is required to
conclude whether both forms are involved in the
catalysis of nitrate reduction (Jormakka, 2004).
The proposed mechanisms in Figure 5 also have
other limitations: it does not take into account of
some Nap structures where there are other ligands
coordinated to Mo in addition to a cysteine. At
Rhodobacter sphaeroides Nap (PDB: 3ML1) Mo-
bis-MGD site, a sulfido group (S
2−
) is doubly bonded
to Mo; for Cupriavidus necator Nap (PDB: 2JIR), the
protein crystal structure shows an additional cyanide
ligand coordinated to Mo. They seem to interfere
with the binding of NO
3
−
, and may also affect the
shifting of oxidation state of the Mo ion. A sulfur
shift mechanism has been proposed to address the
first circumstance (Cerqueira, 2013) (Figure 7). The
Mo-bis-MGD site with a sulfido group is described
as the inactive state, but when a NO
3
−
substrate comes
in, the sulfido ligand will undergo structural changes
to the active state, where a binding site is vacated and
the catalytic reaction can take place as usual.
Figure 7: The sulfur shift mechanism.
Tungsten (W) and Mo are from the same group in
the periodic table, so they share similar chemical
properties, and DMSO reductase (DMSOR) can
function with both Mo and W (Stewart, 2000).
However, nitrate reductases from the DMSO
reductase family, would become inefficient if the Mo
ion of Mo-bis-MGD is replaced by W (Sparacino-
Watkins, 2014). The reason is still unclear, but one
speculation is that W-containing enzymes typically
catalyze reactions with lower redox potentials (E
⦵
≤
−
420
mV) (Moura, 2004). Therefore, they may not
work for the reduction of NO
3
−
whose redox potential
is positive (+
420
mV).
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
508
4.2 Iron–Sulfur Clusters
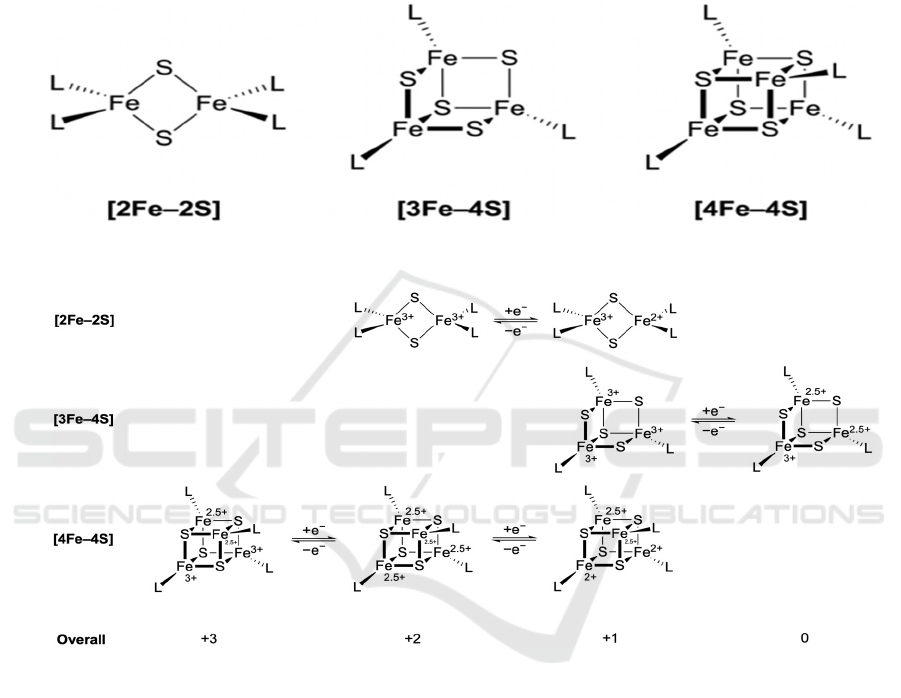
There are three most common iron–sulfur clusters,
respectively [2Fe–2S], [3Fe–4S], and [4Fe–4S]
(Broderick, 2004) (Figure 8a). Iron–sulfur clusters
are ancient structures developed during evolution.
While they are important protein cofactors primarily
responsible for electron transfer, they also have other
functions such as DNA damage repair (Brzóska,
2006). Iron–sulfur clusters transport electrons
through the alteration of Fe oxidation state between
+2 and +3, and since each cluster contains more than
one Fe ion, it may exhibit multiple overall oxidation
states (Beinert, 2000) (Figure 8b).
8a
8b
Figure 8: (a) Three common types of iron–sulfur clusters. L represents any ligand, often cysteine. Though [4Fe–4S] is cubic,
it is not a perfect cube. This is similar for [3Fe–4S]. (b) Electron transfer mechanisms in the three common iron–sulfur
clusters. The oxidation state of +2.5 is due to delocalized electrons, so they occur in pairs.
In prokaryotic nitrate reductases, all of the three
common types of iron–sulfur clusters are present or
possibly present. In NapA, there is a [4Fe–4S] near
the active site (Figures 1 & 9b), which is the same for
NarG. In NarH, the electron transfer subunit, there
are three more [4Fe–4S] and one [3Fe–4S], which is
at the side nearer to NarI, where two heme B are
located (Figure 9a). On the contrary, the
understanding of Nas is still limited and no crystal
structures of Nas have been determined, so it can only
be speculated based on genetic information that there
may be a [4Fe–4S] near Mo-bis-MGD and additional
[2Fe–2S] clusters (Richardson, 2007).
4.3 Hemes
Hemes are primarily composed of porphyrin rings,
with an iron ion chelated by four nitrogen atoms from
porphyrin’s pyrrole rings. Based on different side
chains, hemes are classified into different types, and
the most common ones include heme B and heme C.
Heme B is the precursor of other heme types and it is
not attached to the protein, while heme C is anchored
to the peptide chains through covalent bonding to
thiolate groups of cysteines (Kim, 2018) (Figures 10
& 9c). The iron center of heme is often axially
coordinated by histidine residues (Figure 9c),
Mechanisms and Electron Transfer of Prokaryotic Nitrate Reductases
509
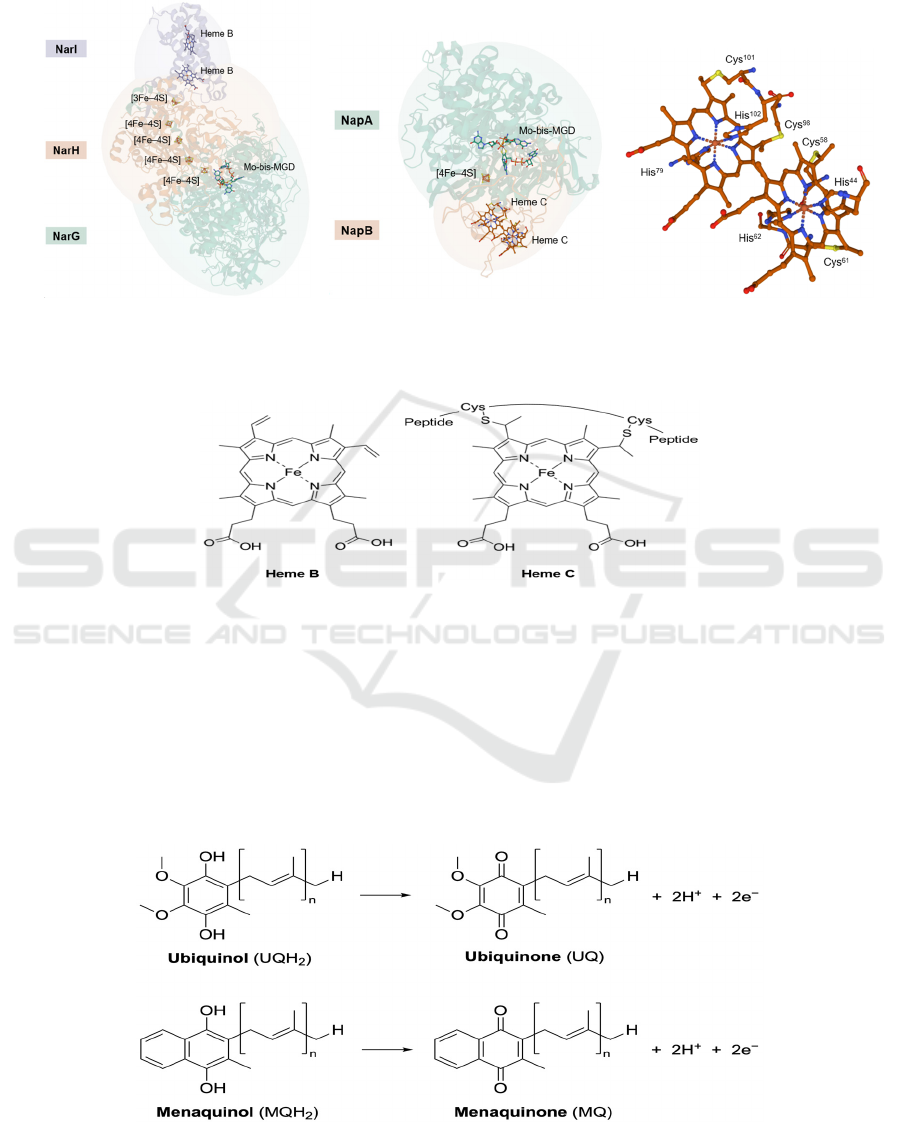
because the protonation state of histidine can tune the
redox potential, spin state and reactivity of the Fe ion
(Bowman, 2008). Since the Fe ion has two oxidation
states, +2 and +3, hemes can also facilitate electron
transfer.
9a 9b 9c
Figure 9: (a) The overall structure of NarGHI of E. coli (PDB: 1Q16). All ligands binding to the labelled cofactors are hided.
(b) The overall structure of NapAB of Rhodobacter sphaeroides (PDB: 1OGY). Ligands binding to heme C are not hided.
(c) A zoom-in view of ligands at heme C. Each heme has two axial histidine ligands.
Figure 10: Structures of heme B and heme C.
Hemes are only present in Nar and Nap, as two
different types. Two heme B are present in NarI and
two heme C are present in NapB. Though crystal
structures of NapC has not been determined, it also
contains heme C. Hemes in NapB only act as electron
carriers, but those in NapC and NarI have another
function, which is to oxidize quinols (including
ubiquinols and menaquinols) to their respective
quinone form, hence generating two electrons
necessary for NO
3
−
reduction (Gates, 2011). For Nar,
this process also releases two protons into the
periplasm, hence creating a proton gradient for ATP
synthesis (Figures 11 & 2). Although the possible
quinol binding sites have been suggested (Bertero,
2005), there are no studies on the mechanism of
quinol oxidation by NapC and NarI.
Figure 11: Chemical equation for oxidation of ubiquinols and menaquinols.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
510

5 ELECTRON TRANSFER IN
NITRATE REDUCTASES
5.1 Membrane-bound Respiratory
Nitrate Reductase (Nar)
To form a complete redox loop and convert quinones
back to quinols, another enzyme is required: formate
dehydrogenase N (Fdn). It is structurally highly
similar to Nar, with three subunits GHI, where FdnG
contains a Mo-bis-MGD and a [4Fe–4S] cluster as
NarG, FdnH contains four slightly different iron–
sulfur clusters compared to NarH, and FdnI anchors
the enzyme on plasma membrane as NarI, and
contains two heme B and Q sites where quinones are
reduced back to quinols (Figure 9a & 12a & 12b).
NarGHI often forms dimers while FdnGHI forms
trimers, and their orientations are different: the FdnG
active site is inside the periplasm (Bertero, 2011)
(Figure 12b).
12a 12b
Figure 12: (a) Crystal structure of E. coli FdnGHI (PDB: 1KQG). The structure shows a trimer, but the cofactors of only one
protein unit are shown. It also shows a quinone molecule at heme B, which is probably the substrate that is going to be
reduced. (b) Simplified representation of the positions of FdnGHI and NarGHI relative to the plasma membrane. Electron
transfers and the Q cycle are also shown.
Figure 13: Distances between each electron transfer cofactor and respective redox potential values for iron–sulfur clusters in
NarH. The values are for E. coli Nar, PDB: 1Q16.
The function of the FdnG active site is to oxidize
formates (HCOO
−
) to carbon dioxide (CO
2
) and a
proton, releasing two electrons which are then
transferred through the [4Fe–4S] clusters in the
electron transfer subunit FdnH to FdnI, where the Q
site is located. Quinones accept the two electrons and
are reduced to quinols, which are then used as the
electron donor for NO
3
−
reduction. In theory, this
process releases two protons into the periplasm, but
it seems that protons produced would be consumed
by quinone reduction at FdnI, and thus conserved in
the Q cycle. Despite this, there is still a net proton
movement from the cytoplasm towards periplasm,
because one proton is produced by the oxidation of
formates while two are consumed in the oxidation of
NO
3
−
.
Mechanisms and Electron Transfer of Prokaryotic Nitrate Reductases
511
Some specific aspects of the route of electron
transfer in Nar are also intriguing. The electron
transfer cofactors are neatly aligned to form a
structure functioning like an electrical wire (Figure
9a), where the distance between adjacent cofactors
are all less than 14 Å, the distance limit for
physiological electron transfer (Bertero, 2011)
(Figure 13). However, the redox potential values
determined in several E. coli Nar iron–sulfur clusters
(Blasco, 2001) are not consistently arranged in a
thermodynamically favorable order, with several
large potential barriers (Figure 13). The reason is still
unclear, but it may involve electron tunnelling (Page,
1999).
5.2 Periplasmic Nitrate Reductase
(Nap)
Instead of iron–sulfur clusters, NapB uses two heme
C molecules to facilitate electron transfer (Figure 9b).
It also has a quinol-oxidizing subunit NapC with two
heme C, whose crystal structure is yet to be
determined. NapC is also anchored to the membrane
(Figure 2).
Unlike Nar, it is believed that amino acid residues
also play a part in the electron transfer pathway in
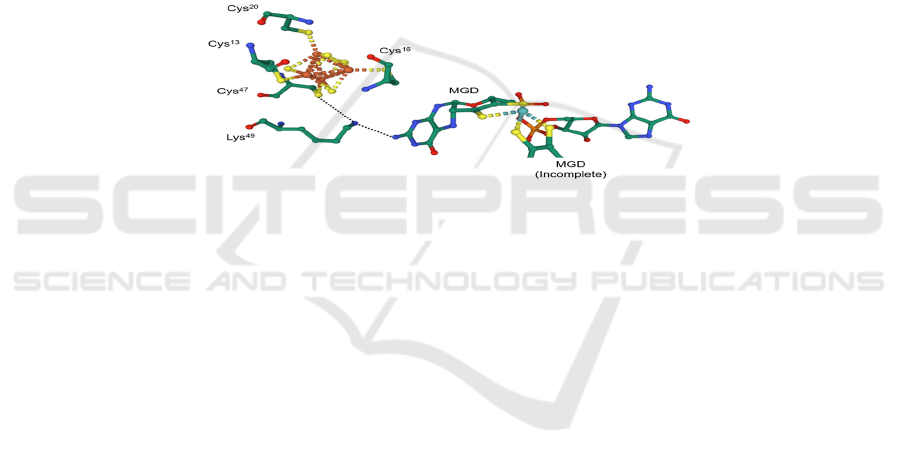
Nap. In NapA, the distance between the Mo ion and
the adjacent [4Fe–4S] cluster is about 12 Å, fairly
close to the 14 Å limit, so a lysine residue which is
hydrogen-bonded to the −NH
2
group of one of the
MGD provides an electron transfer pathway
(Sparacino-Watkins, 2014) (Figure 14). However,
the mechanism is not stated.
Figure 14: The lysine residue (Lys
49
) which facilitates electron transfer in Nap of E. coli (PDB: 2NAP).
5.3 Prokaryotic Assimilatory Nitrate
Reductase (Nas)
Since the major function of Nas is very different from
that of Nar and Nap, its structure also deviates a lot
from Nar and Nap. However, the current
understanding on Nas is rather limited, and no crystal
structures of any subunit of Nas have been
determined.
Nas are free proteins located in the cytoplasm.
They do not use the Q cycle as the electron source to
reduce NO
3
−
, so they do not contain hemes. Instead,
Nas rely on flavodoxin or NADH as the electron
source. Flavodoxin-dependent Nas gains electrons
through the electron carrier flavin mononucleotide
(reduced form, FMNH
2
) via the oxidation FMNH
2
→
FMN + 2H
+
+ 2e
−
and it is composed of a single
subunit, while NADH-dependent Nas has one FAD-
containing subunit receiving electrons from the
oxidation of NADH through NADH → NAD
+
+ H
+
+ 2e
−
, and another catalytic subunit containing Mo-
bis-MGD. Iron–sulfur clusters [4Fe–4S], [3Fe–4S]
and [2Fe–2S] are probably all present for electron
transfer, according to genetic information (Moreno-
Vivián, 1999).
6 CONCLUSIONS
In conclusion, prokaryotic nitrate reductases all use a
Mo-bis-MGD cofactor at the active site to catalyze
nitrate reduction and iron–sulfur clusters to transfer
electrons. Nar and Nap use the Q cycle as the electron
source, so they both contain hemes, while Nas gains
electron through flavodoxin or NADH, where heme
is not required.
There are still lots of problems to solve in the field
of nitrate reductases. The most completely studied
nitrate reductase is Nar, because the crystal structure
of NarGHI is clear, and data about redox potentials
and distances between cofactors have all been
determined. On the contrary, there is little structural
information about Nas, where most are only
speculations. There are also other intriguing aspects
for future studies, such as the potential barrier inside
NarH, the mechanism of quinol oxidation, and why
Mo is the unique element involved in nitrate
reduction.
ICBEB 2022 - The International Conference on Biomedical Engineering and Bioinformatics
512

REFERENCES
Bebout, G. E., Fogel, L. M., & Cartigny, P. (2013).
Nitrogen: Highly Volatile yet Surprisingly Compatible.
Elements, 9(5), 333-338.
Butler, C. S., Charnock, J. M., Bennett, B., Sears, H. J.,
Reilly, A. J., Ferguson, S. J., Garner, C. D., Lowe, D.
J., Thomson, A. J., Berks, B. C., Richardson, D. J.
(1999). Models for Molybdenum Coordination during
the Catalytic Cycle of Periplasmic Nitrate Reductase
from Paracoccus denitrificans Derived from EPR and
EXAFS Spectroscopy. Biochemistry, 38(28), 9000-
9012.
Broderick, J. B. (2004). Iron–Sulfur Clusters in Enzyme
Catalysis. In J. A. McCleverty, & T. J. Meyer,
Comprehensive Coordination Chemistry II: Bio-
coordination Chemistry (Vol. VIII). Elsevier.
Brzóska, K., Męczyńska, S., & Kruszewski, M. (2006).
Iron-sulfur cluster proteins: electron transfer and
beyond. Acta Biochimica Polonica, 53(4), 685-691.
Beinert, H. (2000). Iron-sulfur proteins: ancient structures,
still full of surprises. JBIC Journal of Biological
Inorganic Chemistry, 5(1), 2-15.
Bowman, S. E., & Bren, K. L. (2008). The chemistry and
biochemistry of heme c: functional bases for covalent
attachment. Natural Product Reports, 25(6), 1118-
1130.
Bertero, M. G., Rothery, R. A., Boroumand, N., Palak, M.,
Blasco, F., Ginet, N., Weiner, J. H., Strynadka, N. C.
(2005). Structural and Biochemical Characterization of
a Quinol Binding Site of Escherichia coli Nitrate
Reductase A. Journal of Biological Chemistry,
280(15), 14836-14843.
Bertero, M. (2011). The membrane-bound nitrate reductase
A from Escherichia coli: NarGHI. Encyclopedia of
Inorganic and Bioinorganic Chemistry, 1-10.
Blasco, F., Guigliarelli, B., Magalon, A., Asso, M.,
Giordano, G., & Rothery, R. A. (2001). The
coordination and function of the redox centres of the
membrane-bound nitrate reductases. Cellular and
Molecular Life Sciences, 58(2), 179-193.
Cabello, P., Roldán, M. D., Castillo, F., & Moreno-Vivián,
C. (2009). Nitrogen Cycle. Encyclopedia of
Microbiology, 299-321.
Cerqueira, N. M., Fernandes, P. A., Gonzalez, P. J., Moura,
J. J., & Ramos, M. J. (2013). The Sulfur Shift: An
Activation Mechanism for Periplasmic Nitrate
Reductase and Formate Dehydrogenase. Inorganic
Chemistry, 52(19), 10766-10772.
Dias, J. M., Than, M. E., Humm, A., Huber, R., Bourenkov,
G. P., Bartunik, H. D., Bursakov, S., Calvete, J.,
Caldeira, J., Carneiro, C., Moura, J. J., Moura, I.,
Romão, M. J. (1999). Crystal structure of the first
dissimilatory nitrate reductase at 1.9 Å solved by MAD
methods. Structure, 7(1), 65-79.
Gates, A. J., Kemp, G. L., To, C., Mann, J., Marritt, S. J.,
Mayes, A. G., Richardson, D. J., Butt, J. N. (2011). The
relationship between redox enzyme activity and
electrochemical potential—cellular and mechanistic
implications from protein film electrochemistry.
Physical Chemistry Chemical Physics, 13(17), 7720-
7731.
Jormakka, M., Richardson, D., Byrne, B., & Iwata, S.
(2004). Architecture of NarGH Reveals a Structural
Classification of Mo-bisMGD Enzymes. Structure,
12(1), 95-104.
Kuypers, M. M., Marchant, H. K., & Kartal, B. (2018). The
microbial nitrogen-cycling network. Nature Reviews
Microbiology, 16(5), 263-276.
Kim, H. J. (2018). Haem Structure and Function. eLS, 1-9.
Lippard, S. J., & Berg, J. M. (1994). Principles of
Coordination Chemistry Related to Bioinorganic
Research. In S. J. Lippard, & J. M. Berg, Principles of
Bioinorganic Chemistry. University Science Books.
Moreno-Vivián, C., Cabello, P., Martinez-Luque, M.,
Blasco, R., & Castillo, F. (1999). Prokaryotic Nitrate
Reduction: Molecular Properties and Functional
Distinction among Bacterial Nitrate Reductases.
Journal of Bacteriology, 181(21), 6573-6584.
Moura, J. J., Brondino, C. D., Trincão, J., & Maria João, R.
(2004). Mo and W bis-MGD enzymes: nitrate
reductases and formate dehydrogenases. JBIC Journal
of Biological Inorganic Cheistry, 9(7), 791-799.
Page, C. C., Moser, C. C., Chen, X., & Dutton, P. (1999).
Natural engineering principles of electron tunnelling in
biological oxidation–reduction. Nature, 402(6757), 47-
52.
Richardson, D. J., van Spanning, R. J., & Ferguson, S. J.
(2007). The Prokaryotic Nitrate Reductases. In H.
Bothe, S. Ferguson, & W. E. Newton, Biology of the
Nitrogen Cycle. Elsevier.
Sparacino-Watkins, C., Stolz, J. F., & Basu, P. (2014).
Nitrate and periplasmic nitrate reductases. Chem. Soc.
Rev., 43(2), 676-706.
Stewart, L. J., Bailey, S., Bennett, B., Charnock, J. M.,
Garner, C. D., & McAlpine, A. S. (2000).
Dimethylsulfoxide reductase: an enzyme capable of
catalysis with either molybdenum or tungsten at the
active site. Journal of Molecular Biology, 299(3), 593-
600.
Mechanisms and Electron Transfer of Prokaryotic Nitrate Reductases
513
