
Post-market Clinical Follow-up (PMCF) GAP Analysis for
Legacy Devices Class III between
the Medical Device Directive (MDD 93/42/EEC) and
the Medical Device Regulation (MDR 2017/745)
Marina Makeenko
1
and Thierry Chevallier
2,3,4
1
Biotechni SAS, 178 Avenue du Serpolet, La Ciotat, France
2
Department of Biostatistics, Epidemiology, Public Health and Innovation in Methodology (BESPIM),
CHU Nîmes, Place du Pr. Robert Debré, 30029 Nîmes, France
3
UMR 1302, Institute Desbrest of Epidemiology and Public Health, INSERM, Univ. Montpellier, Montpellier, France
4
Tech4Health-FCRIN, France
Keywords: Medical Device, Post-market Clinical Follow-up, PMCF, GAP Analysis, Legacy Devices.
Abstract: The passage from the MDD 93/42/CEE to the MDR 2017/745 remains a big challenge for the manufactures.
The interpretation of the regulatory requirements stays unclear and can differ from one source to another,
especially when it comes to the clinical evaluation. Will the data collected under the MDD 93/42/CEE be
sufficient to prove the safety and security of the device? Under the directive each country was establishing its
own requirements for the conduct of the studies. The MDR has standardized these rules, so that all the clinical
data collections follow the same pathway. We will examine the PMCF of the class III devices already CE
marked under the directive (legacy devices) to find out if the new requirements will be asked to be in
compliance with the MDR. A Gap analysis between the MDD and MDR will help us in our research. A matrix
in the form of a questionnaire will be established to help us verify compliance of the PMCF under the MDR.
1 INTRODUCTION
The Medical Devices Regulation 2017/745 (MDR)
came in force on the 26th of May 2021 bringing
significant regulatory changes.
The new MDR requirements reinforced clinical
data, technical documentation, and labelling.
However, the most significant change concerned the
clinical part, as the manufacturers have to obtain a
bigger clinical data to prove safety and performance
of their products.
As the representative of the medium size
company, manufacturing implantable medical
devices class III we are at the heart of regulatory
constraints, which are becoming more and more
imposing. Our products already have a long
marketing history; therefore, they enter in the
category of legacy device.
“Legacy devices are all devices previously CE
marked under the European Medical Devices
Directive 93/42/EEC” (MDCG 2020-6).
We will analyse the requirements for the PMCF
report under MDR 2017/745 to build the gap analysis
between MDD and MDR, paying special attention to
the interpretation of the meaning « sufficient clinical
data », since this essential requirement of the MDR is
not clearly explained.
We will rely our researches on the MDR, Medical
Device Coordination Group (MDCG) and MEDDEV
guidelines. A literature review will be done using
scientific publications, notify body and consultancy
agencies articles.
We will apply our research on the example of a
Biotechni S.A.S., a family-owned company created in
1984 in Marseille and currently employing 48
members. Biotechni designs, producers and
commercialises a range of implants for hip, shoulder
and spine (classes I, IIa, IIb and III). All the products
produced by Biotechni are marketed since at list 10
years and are covered by a valid certificate issued in
accordance with Directive 93/42/EEC, valid until
May 2024.
To illustrate our gap analysis, we will use an
example of a femoral stem, used in association with a
femoral head and acetabular cup for a hip joint
replacement.
Makeenko, M. and Chevallier, T.
Post-market Clinical Follow-up (PMCF) GAP Analysis for Legacy Devices Class III between the Medical Device Directive (MDD 93/42/EEC) and the Medical Device Regulation (MDR
2017/745).
DOI: 10.5220/0010889400003123
In Proceedings of the 15th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2022) - Volume 1: BIODEVICES, pages 273-280
ISBN: 978-989-758-552-4; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
273

Below is the brief description of the device in
question.
Table 1: Filler-3ND femoral stem brief description.
MANUFACTURER BIOTECHNI S.A.S
PRODUCT RANGE FILLER-3ND® 135°
Titanium cementless femoral
hip stems (11 sizes)
PICTURE
CLASS III
INTENDED PURPOSE For use in total and partial
hip arthroplasty
Date of 1st CE marking 25/03/2004
TOTAL SALES 18 000
2 CLINICAL REQUIREMENTS
FOR LEGACY DEVICE
“Past performance is no guarantee of future results.
Study Proves Past Results Doesn’t Predict Future
Results…”
The founding fathers of the new MDR were
obviously very inspired by those quotes while
building the new regulations.
Therefore, even if the device has been marketed
for decades with no significant change in design, it
doesn’t exempt the manufacturer from the
complimentary clinical studies.
2.1 Exemptions from the Clinical
Requirements for Legacy Device
According to MDR Article 61(4 and 6) clinical
investigations shall be performed for Class III and
implantable devices, except if they have been
previously marketed under Directive and their
clinical evaluation is based on sufficient clinical data.
Another exemption from clinical investigations is
given in MDR Annex XIV, Part A (3), where it is
stipulated, that a clinical evaluation may be based on
clinical data relating to a device for which
equivalence to the device in question can be
demonstrated by technical, biological and clinical
characteristics with the authorisation of the full access
to the technical documentation.
2.2 Sufficient Clinical Data
Different sources are attempting to explain the
meaning of the word « sufficient ».
The Article 61(1) of the MDR states that
: “The
level of clinical evidence shall be appropriate in view of the
characteristics of the device and its intended purpose.”
Section 4 of MDCG 2020-6 guidance states:
“Both
the Directives and the MDR require the quantity and quality
of clinical data to be sufficient to demonstrate safety,
performance and the acceptability of the benefit-risk
ratio…and require clinical evidence to be sound and the
conclusions derived from this evidence to be scientifically
valid.”
Section 5 of the guidance tries to explain what
does the word sufficient means by saying:
“sufficient
clinical evidence is understood as the present result of the
qualified assessment which has reached the conclusion that
the device is safe and achieves the intended benefits.”
Does it really help to understand what does
“sufficient” mean? We are not so sure…
The manufacturer should conduct an analysis to
determine if additional data or change in PMCF
design to support the clinical evidence are required to
meet additional MDR requirements. This could be
achieved through a gap analysis with respect to new
MDR requirements.
The gap analysis is the difference between what
we have with MDD and what we should have to
comply with MDR.
To prepare the GAP analysis of the PMCF report
in the most exhaustive way we decided to separate our
researches in 3 main pillars:
1. Acceptable quantity of clinical data
2. Acceptable quality of clinical data
3. Additional sources of clinical and not clinical
data
The acceptable quantity of clinical data is the
quantity of information we need to make the study
results reliable and representing of a real life.
The acceptable quality of clinical data will be
appraised by its methodological quality and its
relevance.
The additional sources of clinical and not clinical
data will be considered to implement the PMCF
results.
The goal is to establish a generic matrix that could
be used for each legacy medical device.
ClinMed 2022 - Special Session on Dealing with the Change in European Regulations for Medical Devices
274

2.3 Acceptable Quantity of Clinical
Data
2.3.1 Sample Size
The choice of the sample size is one of the primary
endpoints the sponsor has to determine for the clinical
study.
As we can’t include all the population of interest,
we should determine what is the minimum number of
patients that would reflect as much as possible the
total population of interest, and therefore make study
results statistically significant.
The MDR requires to document the choice of
sample size, and to provide a rationale explication of
the procedures and the methods used.
The sponsor should define what endpoints would
be statistically measured through the clinical study to
demonstrate the device general safety and
performance conformity assessment. To assess
quality and performance of an implantable medical
device the survival rate is usually measured.
2.3.2 Duration of Clinical Study
The duration of a clinical investigation is also crucial
and must be considered while planning the PMCF.
“The follow-up period during the clinical investigation
shall permit the demonstration of clinical performance,
effectiveness or safety over a period of time sufficient to
represent a realistic test of the investigational device and
allow any risks associated with adverse device effects to be
identified and assessed” (ISO 14155 : 2020).
“Although there is not enough information yet available
to calculate exactly how long a hip replacement will last,
using available arthroplasty registry data, we estimate that
about three-quarters of hip replacements last 15–20 years
and just over half of hip replacements last 25 years in
patients with osteoarthritis” (How long does a hip
replacement last? A systematic review and meta-analysis of
case series and national registry reports with more than 15
years of follow-up - Jonathan T Evans, Jonathan P Evans,
Robert W Walker, Ashley W Blom, Michael R
Whitehouse*, Adrian Sayers* - 2019).
Taking into consideration the average duration of
the hip joint life cycle and the common practice in
designing the hip replacement PMCF studies, we
estimated the minimum duration of the study at 10
years.
2.4 Acceptable Quality of Clinical Data
“Clinical investigation that are currently being conducted
with respect to Directive 93/42/EC and Directive
90/385/EC by the date of application of the MDR, can
continue to be conducted” (MDCG 2021-6).
Therefore, the Clinical investigation protocols
that have been approved under Directive 90/385/EC
can be keep going, as long as the quantity and quality
of the data are sufficient. But what constitutes the
quality of the data?
The quality appraisal of the clinical data is
uncertain, as it has to take into consideration several
aspects.
MEDDEV 2.7/1 rev. 4. point 9. suggests to
evaluate two main sources: the methodological
quality of the data, and the relevance of the data.
2.4.1 Methodological Quality
Different methods are available to conduct a clinical
study.
In case of legacy device observational prospective
or retrospective studies are most commonly used.
We are encouraged to conduct the methodology
evaluation to assess whether there are any points that
can be improved.
We have grouped the information from Appendix
A6 General principles of clinical evaluation of
MEDDEV 2.7/1 rev. 4. to constitute the checklist of
the desired parameters for a high-level scientific
validity study to demonstrate adequate clinical
performance and clinical safety.
Below are the points that should be taken into
consideration while assessing the methodological
quality of the study:
- Sufficient information on elementary aspects
- Proper statistical methods
- Adequate controls
- Proper collection of mortality and serious
adverse events data
- Legal activities
- Schedule for PMCF activities
Let’s see in details those points.
- Sufficient Information on Elementary Aspects
The clinical data should necessarily contain the
following elementary aspects:
a) Methods used
b) Products used
c) Number of patients
d) Clinical outcomes
e) Undesirable side-effects
f) Confidence intervals/ calculation of statistical
significance
g) Reference to the harmonised standards or
guidances.
Post-market Clinical Follow-up (PMCF) GAP Analysis for Legacy Devices Class III between the Medical Device Directive (MDD
93/42/EEC) and the Medical Device Regulation (MDR 2017/745)
275
- Adequate Controls
a) Objective control parameters
b) Assessed endpoints are not subject to natural
fluctuations
c) No other treatments, that can influence the
clinical outcome are taken
d) Any other influencing factors
- Proper Collection of Mortality and Serious
Adverse Events Data
The lost to follow-up should be avoided as much as
possible. Therefore, the Investigator should have a
contact person that will be contacted if a patient is lost
to follow-up. The emergency contact should be
provided while recruitment. It could be mentioned in
the PMCF protocol.
The Sponsor should be immediately informed
about all adverse events or any sort of failures.
- Legal Activities
MDR clinical investigation requirements are based on
existing ethical and legal regulations.
“Clinical
investigations should be in line with well-established
international guidance in this field, such as the international
standard ISO 14155:2011” (Point 64 of the preamble to
MDR).
Article 74 precises that rules provided in points
(b) to (k) and (m) of article 62(4), article 75, article
76, article 77, article 80(5) and (6) and the relevant
provisions of Annex XV shall apply to all PMCF
investigations. This must be understood, that both
PMCF investigations with invasive or burdensome
procedures as well as the investigations free of such
additional measures should comply with the same
requirements.
Within MDR it must be understood that in-label,
observational, non-interventional studies will
however stay in a general category of clinical
investigation. That means that all the requirements
and obligations set up for the PMCF investigation
within the framework of the MDR must be observed.
The fact that the studies stay in the scope of
intended purpose and without bringing any additional
risk to patients does not exempt them from general
obligations.
Below is the list of the essential requirements to
the clinical investigation according to the ISO14155:
2020, and the analysis of this new requirements
applied to the Filler-3ND observational retro-
prospective study, started under MDD.
As we can see the design study started under
MDD can be improved and adjusted to comply as
much as possible with the new requirements.
Table 2: List of essential requirements ISO14155:2020
applied to Filler-3ND PMCF report.
List of
requirements
ISO14155-2020
Filler-3ND
PMCF report
Comments
Investigation
brochure
No Can be added
Clinical
Investigation Plan
Yes (PMCF
protocol)
Principle
Investigators CV
No Can be added
List of
investigation sites
No Can be added
Ethics committee
approval
For certain
countries
Ask for a EC
approval if a
new country
included
Regulatory
authorities’
approval
Yes
Signed agreement
between
investigator and
sponsor
Yes
Financial
agreement between
investigator and
sponsor - Yes
Yes
Insurance No
As the study
started under
MDD no need
to include it a
posteriori
Investigation site
selection report
No Can be added
- Schedule for PMCF Activities
Annex XIV, part B, point 6.2 (h) recommends
“a
detailed and adequately justified time schedule for PMCF
activities (e.g., analysis of PMCF data and reporting) to be
undertaken by the manufacturer”
.
PMCF report should indicate the targeted study
duration for subjects already enrolled or/and to be
enrolled to achieve target sample size.
- Conclusion About Study Design
We analysed point by point all the aspects, that
constitute the methodological quality of a study.
But as mentioned earlier, the quality of the study is
based on 2 pillars: the methodology and the relevance
of the data. In the next chapter we will analyse the
second pillar of the quality assessment, what
constitutes the data relevance of the study.
ClinMed 2022 - Special Session on Dealing with the Change in European Regulations for Medical Devices
276

2.4.2 Relevance of the Data
Article 61 of the MDR specifies the general
requirements regarding clinical investigations to
demonstrate conformity of devices.
According to article 62 of the MDR the clinical study
should demonstrate, that the following points have
been achieved:
1. Device achieves the performance intended as
specified by its manufacturer
2. Establish and verify the clinical benefits of a
device as specified by its manufacturer
3. Establish and verify clinical safety, detect any
undesirable side effects
4. Evaluate the benefit / risk ratio
All that information claimed by the manufacturer
should be confirmed by clinical data.
- Conformity with the IFU Allegations
Annex I, CHAPTER III points 23.4 of the MDR
precises the information that the manufacturer should
supply in the instructions for use.
The assessor will examine whether there is sufficient
clinical evidence to demonstrate that the device
performs as intended in the IFU.
- Benefits Claimed in Marketing Material
The information given about a medical device in the
marketing material must be consistent with the
manufacturer's intended purpose and the scope of use
of the medical device. Moreover, the allegations
claimed by the manufacturer in the advertising
information must be supported by clinical evidence.
- Target Groups
The target population should be identified in the
PMCF through the inclusion and exclusion criteria.
The inclusion/exclusion criteria must be strictly
respected as it can compromise the clinical results.
Generally, the population included in the study
should be homogenous which benefits from the same
level of infrastructures. Investigation site selection
report can be helpful to involve the centers of the
same level. The investigators should have the same
knowledge as well. Training records, providing
evidence, that the investigators have been trained are
very helpful.
3 ADDITIONAL SOURCES OF
CLINICAL AND
NON CLINICAL DATA
Annex XIV, part B, point 6.2 (a) of MDR precises
what are the available sources of the PMCF data
collection:
“the general methods and procedures of the
PMCF to be applied, such as gathering of clinical
experience gained, feedback from users, screening of
scientific literature and of other sources of clinical data”
.
Therefore, feedback from users and screening of
scientific literature can be put in place to implement
the PMCF.
In fact, user surveys are listed in the MDR as a
valid method of post-market clinical data collection.
3.1 Market Experience Feedback
PMCF surveys should not be ignored, as they
represent many advantages.
With the PMCF surveys the clinical data can be
significantly implemented.
The PMCF survey should have a clear objective
and not have conflicting purposes that could confuse
the PMCF results. PMCF surveys should be in line
with the PMCF clinical data collection and reply to
the same objectives, as precised in the Annex XIV,
part B, point 6.1.
The regular collection of the feedback from users
may help to identify any unknown side-effects,
emergent risks, misuse or off-label use of the device.
3.2 Alternate Therapies/State of the
Art/Current Knowledge
A critical review of the literature while assessing the
state of the art, alternative examination and treatment
methods should be considered when implementing
the PMCF.
The literature search should demonstrate if the
device in question is a well-established practice with
numerous articles validating both design, longevity
and security.
If the device is identified as belonging to the group
of « well-established technologies » a lower level of
clinical evidence may be justified to be sufficient for
the confirmation of conformity with relevant GSPRs.
3.3 Benefit-Risk Analysis
The aim of the PMCF plan is ensuring the continued
acceptability of the benefit-risk ratio, referred to in
Section 1 and 9 of Annex I in the MDR.
Post-market Clinical Follow-up (PMCF) GAP Analysis for Legacy Devices Class III between the Medical Device Directive (MDD
93/42/EEC) and the Medical Device Regulation (MDR 2017/745)
277

The benefit-risk analysis should be documented in
the clinical evaluation report using all the available
sources of clinical and non-clinical data, obtained
through the PMCF activities.
Aspects that influence the acceptability of benefits
and risks can be found in Appendix A7.2 and in
Appendix A7.4 of MEDDEV 2.7/1 rev. 4.
The information gathered through the PMCF
activities should be sufficient to confirm the
acceptable benefit-risk ratio.
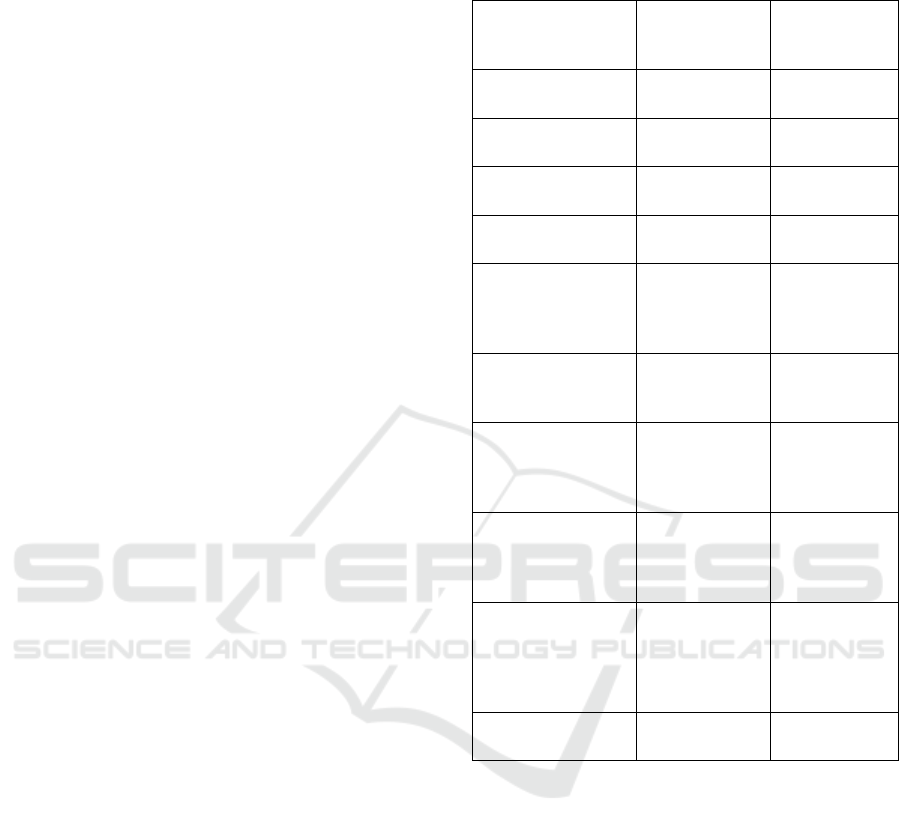
3.4 Gap Analysis Matrix
The main goal of this thesis was to establish a check
list, that will help to conduct a gap analysis of a
PMCF report. We grouped all the collected
information in a table-matrix.
We have illustrated this matrix with the example
of the Filler-3ND PMCF report.
Table 3: GAP analysis Matrix.
Product Name
Product Class
Q.N° Question Response
Gaps, if
any/recom-
mendation
Sample size
Q1 What is the target size of the sample?
Q2 Has the sample size calculation been justified?
Q3 Has the sample size justification been approved by the Notify Bod
y
?
Q4 Has the target sample size
b
een reached?
Duration of the study
Q5 Is the duration of the study enough to demonstrate its safety and performance?
Methodology
Elementary aspects
Q6 Does the PMCF disclose the methods used?
Q7 Does the PMCF disclose the
p
roducts used?
Q8 Does the PMCF disclose the number of patients included?
Q9 Does the PMCF disclose all the clinical outcomes?
Q10
Does the PMCF disclose data from others activities? (device registry, PMCF studies, real world
evidence, surveys about the use of device, etc…)
Q11 Have the undesirable side-effects been observed?
Q12 Have confidence intervals/ calculation of statistical significance been used?
Q13 Does the study PMCF reference to any harmonised standards, relevant guidance on PMCF
Adequate controls
Q14 Are the endpoin
t
s assessment objective? (ex., pain is subjective)
Q15
Are the endpoints or symptoms assessed are subject to natural fluctuations? (ex. when the natural
evolution of the pathology is not clearly predictable)?
Q16 Are any other treatments, that can influence the clinical outcome taken?
Q17
May clinical outcomes can be affected by variability of the patient population, of the disease, of user
skills, of infrastructure…?
Collection of mortality and serious adverse events data
Q18 Has the consent of the subjects for contacting reference persons been obtained?
Q19 Is any failure or adverse event immediately reported?
Legal activities
Q20 Does the study have Investigation brochure?
Q21 Does the study have Clinical Investigation Plan?
Q22 Does the study have Principle Investigators CV?
Q23 Does the study have List of investigation sites?
Q24 Does the study have Ethics Committee approval?
Q25 Does the study have Regulatory Authori
t
y approval?
Q26 Does the study have signed agreement between investigator and sponsor?
Q27 Does the study have financial agreement between investigator and sponsor?
Q28 Does the study have insurance?
Q29
Does the study have Investigation site selection report (verifies that the qualification of investigation
site members has
b
een approved)?
Q30 Does the study have CRF?
Q31 Does the study have Adverse events form?
Q32 Does the study have Device deficiency form?
Q33 Does the study have Training records (evidence, that the investigators have been trained)?
ClinMed 2022 - Special Session on Dealing with the Change in European Regulations for Medical Devices
278

Table 3: GAP analysis Matrix (cont.).
Product Name
Product Class
Q.N° Question Response
Gaps, if
any/recom-
mendation
PMCF Schedule
Q34 Is there a PMCF Follow-Up schedule for on-going subjects?
Q35 Is there a PMCF Follow-Up schedule for additional planned subjects?
Relevance of the data
Indication/intended use/intended purpose in IFU
Q36 Is the exact indication/intended use/intended purpose as described in the device's IFU captured?
List down the indications/intended use/intended purpose in the below rows as per the IFU.
Q37 Indication # 1
Q38 Indication # 2
Benefits claimed in marketing material
Q39 Are the claims as described in the marketing material captured? List down the claims in the below
rows as per marketing material.
Q40 Claim # 1
Q41 Claim # 2
Contraindications/ Warnings & Cautions / Risks in IFU
Q42 Are the exact contraindications as described in the device's IFU captured?
Q43 Are the exact Warnings & Cautions as described in the device's IFU captured?
Q44 Are the exact Risks as described in the device's IFU captured?
Q45 Are previously unknown side-effects identified?
Q46 Are emergent risks identified and analysed?
Q47 Do the clinical results meet the expected benefits?
Q48 Is the benefi
t
-risk ratio continuously acceptable?
Q49 Is the performance characteristics of the device demonstrated?
Target group(s)
Q50 Has the inclusion/ exclusion criteria been respected by the target population?
Q51 Are the infrastructures of the sample homogeneous?
Q52 Is the social and economic level of the sample homogeneous?
Q53 Is the morphology of the sample homogeneous?
Additional sources of clinical and not clinical data
Market Experience feedback
Q54 Has market experience data been collected (complaints, medical device reports, customer surveys,
etc)? List down the data, that has been collected from the market.
Q55 Market experience data # 1
Q56 Market experience data # 2
Q57 Are possible systematic misuse or off-label use of the device identified?
Q58 Are the usability forms filled in regularly?
Critical analysis of the literature
Q59 Is a thorough literature search performed to identify state of the art therapy/management/diagnostic
options available?
Q60 Is a description of all the available therapeutic/management/diagnostic options, historical context
and developments included?
Q61 Is a thorough literature search performed to identify state of the art therapy/management/diagnostic
options available?
Benefit-risk Analysis
Q62 Are all the risks identified from different sources?
Q63 Are the risks acceptable according to current knowledge/ the state of the art in the medical fields
concerned and according to available medical alternatives?
Q64 Is justification available for acceptability of risk(s)?
Q65 Are there any new risk(s) identified?
Q66 If new risks are identified, is the available clinical data sufficient to verify that the device is in
conformity with all the essential requirements pertaining to clinical performance and clinical
safety?
Q67 Does the risk/benefit analysis summarized, considering the current knowledge/the state of the art?
Q68 Does the report explain why the benefit/risk profile and the undesirable side-effects are acceptable
in relation to current knowledge/the state of the art?
Post-market Clinical Follow-up (PMCF) GAP Analysis for Legacy Devices Class III between the Medical Device Directive (MDD
93/42/EEC) and the Medical Device Regulation (MDR 2017/745)
279
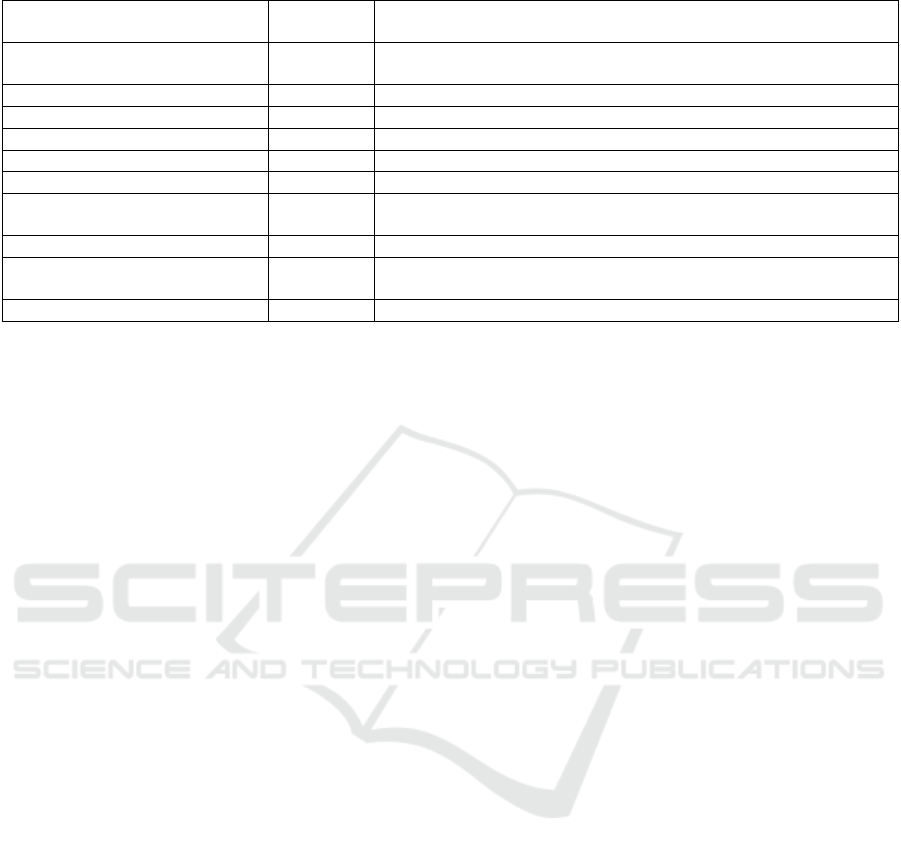
Table 4: Non-compliances of the Filler-3ND PMCF report, detected by GAP analysis Matrix.
GAP Endpoint Level of
importance
Correction action
Collection of mortality and
serious adverse events data
Medium Include reference persons to contact in case of lost to follow-up
durin
g
the recruitment
Le
g
al activities Mino
r
To add Investi
g
ation brochure
Mino
r
To add Principle Investigators CV
Mino
r
Ask for a EC approval if a new country include
d
Insurance Mino
r
Not possible to ad
d
Mediu
m
To add Investi
g
ation site selection re
p
ort
Medium To add training records (evidence, that the investigators have been
trained)
Indication/intended use in IFU Mediu
m
Precise « total or
p
artial re
p
lacement » in the CRF
Target groups Medium Include training records, providing evidence, that the investigators
have been traine
d
Market Experience feedbac
k
Mediu
m
Market feedback should be collected at list once a yea
r
4 CONCLUSION/DISCUSSIONS
MDD to MDR transition is a very challenging step,
and the key of success is the organization. The
establishment of the regulatory roadmap with
deadlines and budgeting is absolutely necessary.
GAP analysis for all the important endpoints could be
very helpful. In our thesis we assessed PMCF report,
but other topics could be reviewed:
- CE Marking Technical File or Design Dossier
- Current device class and product families
- Risk management file review
- Clinical Evaluation Report(s)…
Below is the table that resume all the identified non-
compliances, detected by the GAP Analysis of the
Filler-3ND femoral stem. We estimated the level of
each non-compliance and suggested possible
correction action.
We can see that the PMCF Gap Analysis results
are good and some non-compliances can easily be
corrected, except one, but it is a minor non-
compliance.
We are convinced that the sufficient clinical data
is the most important point of the new MDR. It is
necessary to collect the data systemically in the most
exhaustive way.
The key success in this process is the investigators
implication. Therefore, our goal is to instill in
investigators the importance of clinical follow-ups,
by bringing arguments and following them in each
step of data collection.
REFERENCES
Regulation (EU) 2017/745 of the European parliament and
of the council of 5 april 2017.
Directive 93/42/CEE du conseil du 14 juin 1993 relative
aux dispositifs médicaux.
ISO 14155 – Clinical investigation of medical devices for
human subjects – Good clinical practice.
Guideline for good clinical practice E6(R2), 23 July 2015.
MEDDEV 2.7/1 revision 4 - Clinical evaluation: a guide for
manufacturers and notified bodies under directives
93/42/EEC and 90/385/EEC.
MEDDEV 2.12/2 rev2 – Post market clinical follow-up
studies - a guide for manufacturers and notified bodies.
Guide methodologique HAS – Choix méthodologiques
pour le développement clinique des dispositifs
médicaux, 4 octobre 2013.
MDCG 2020-6 – Regulation (EU) 2017/745: Clinical
evidence needed for medical devices previously CE
marked under Directives 93/42/EEC or 90/385/EEC.
How PMCF surveys can help collect clinical evidence
remotely during the COVID-19 pandemic.
MHRA guidance for manufacturers, 2020.
How long does a hip replacement last? A systematic review
and meta-analysis of case series and national registry
reports with more than 15 years of follow-up - Jonathan
T Evans, Jonathan P Evans, Robert W Walker, Ashley
W Blom, Michael R Whitehouse*, Adrian Sayers* -
2019.
ClinMed 2022 - Special Session on Dealing with the Change in European Regulations for Medical Devices
280
