
A Rare Case of Epidermolytic Hyperkeratosis: Recognition of
Distinctive Clinical and Histopathological Signs
Sarah Mahri
1*
, Lidwina Anissa
1
, Rinadewi Astriningrum
1
, Githa Rahmayunita
1
,
Triana Agustin
1
, Rahadi Rihatmadja
1
1
Department of Dermatology and Venereology Faculty of Medicine Universitas Indonesia/
Dr. CiptoMangunkusumo National Central General Hospital, Indonesia
Keywords: Epidermolytic hyperkeratosis, clinical signs, histopathology
Abstract: Epidermolytic hyperkeratosis (EHK) is a rare autosomal dominant genodermatosis with a prevalence of
1:100,000 to 1:300,000.Mutations primarily of keratin 1 or keratin 10 cause defective keratinization, leading
to skin fragility, blistering, and hyperkeratosis. Neonates with EHK are at risk of developing electrolyte
imbalance, sepsis and malnutrition leading to a considerable mortality. Therefore its diagnosis is important.
As the clinical features of EHK become more apparent with age, a wide spectrum of other genodermatosis
should be considered as differentials at different stages of the disease process. A 5-year-old boy presented to
our department with dirty brown, corrugated plaques distributed all over his body. He had had history of
trauma-related blistering since two days after birth. As he aged, there was a decrease in development of
blisters and erosions, with accompanying increase in severity of hyperkeratosis and foul odor. Physical
examination revealed thickened, brown plaques over the neck, trunk, extremities, and scalp. Cobblestone
pattern were visible over the knees, elbows, and posterior of hands and feet, in addition to multiple
superficial erosions. Histopathologic examination showed massive hyperkeratosis, acanthosis, spongiosis,
lysis and clumping of keratinocytes in the stratum spinosum to granulosum. The diagnosis of EHK was
made. Vaseline, coconut oil, and antiseptic soap gave slight, but acceptable improvement. EHK is rare, thus
recognizing its distinctive clinical patterns is necessary to avoid delayed diagnosis and gave necessary
genetic counselling and prompt treatment.
1 INTRODUCTION
Epidermolytic hyperkeratosis (EHK), also known as
bullous congenital ichthyosi form erythroderma is an
autosomal dominant trait with a prevalence of
approximately 1 in 100,000 to 300,000 persons.
1-
4
The disease is named for the distinctive
histopathologic feature of vacuolar degeneration and
associated hyperkeratosis of the epidermis.
4
EHK is caused by mutations in keratin 1 or keratin
10, resulting in subsequent skin cell collapse and
fragility. This clinically manifests as blistering, later
followed by hyperkeratosis and hyper proliferation.
Onset typically occurs in newborn, with generalized
erythroderma. Erosions, blisters, and peeling are
present. Although the blistering and erythema often
improve over time, hyperkeratotic scale becomes
prominent, most commonly over the neck, hands,
feet, and joints. Other affected areas include the
scalp and infragluteal folds.
1-4
Neonates with EHK
are at risk of developing sepsis,electrolyte
imbalance, and malnutrition leading to a
considerable mortality, therefore its diagnosis is
important.
Between 2016-2018 there were no cases of
epidermolytic hyperkeratosis in our institution. We
herein report a rare case of epidermolytic
hyperkeratosis in a 5-year-old boy. The purpose of
this report is to familiarize with its characteristic
features that enable clinician to make early diagnosis
and distinction with other ichthyosis and bullous
diseases of childhood, as well as to discuss the
therapeutic possibilities.
2 CASE
A 5-year-old boy was consulted to our clinic with
dirty brown, corrugated hyperkeratotic plaques
Mahri, S., Anissa, L., Astriningrum, R., Rahmayunita, G., Agustin, T. and Rihatmadja, R.
A Rare Case of Epidermolytic Hyperkeratosis: Recognition of Distinctive Clinical and Histopathological Signs.
DOI: 10.5220/0009985902570261
In Proceedings of the 2nd International Conference on Tropical Medicine and Infectious Disease (ICTROMI 2019), pages 257-261
ISBN: 978-989-758-469-5
Copyright
c
2020 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
257
distributed all over his body beginning two years
before.
The history started two days after birth, when he
developed blisters on his trunk and extremities,
which ruptured and became eroded. Lesions recurred
over his whole body, sparing the palms, soles, and
face. Blisters and erosions usually healed leaving
hyperpigmentation without scarring. Although he
continued to develop erythema and blisters, they
decreased in severity and frequency with age.
Superficial erosions ceased few months after his
birth and followed by hyperkeratosis lesions.
By the age of three, the patient developed
generalized hyperkeratosis and scaling over his
trunk, extremities and scalp. The lesions
occasionally became pruritic. He would scratch and
peel the affected skin and sometimes they became
infected. As hyperkeratosis became more
pronounced, so did the foul odor. The disease did
not appear to involve the nail or impair neurological
functions.
He has a history of hyper IgE and delayed
speech. He is the only child of healthy, non-
consanguineous parents. No family history of similar
disorders and no history of restrictive membrane at
birth.
Thickened, brown, dirty corrugated plaques were
found on physical examination, distributed over the
neck, trunk, extremities, and scalp, affecting
approximately 95% of his body surface area.
Cobblestone pattern were visible over the knees,
elbows, and posterior of hands and feet. Areas of
superficial erosion were evident on his left ear, back,
upper and lower limbs. His scalp hair were enclosed
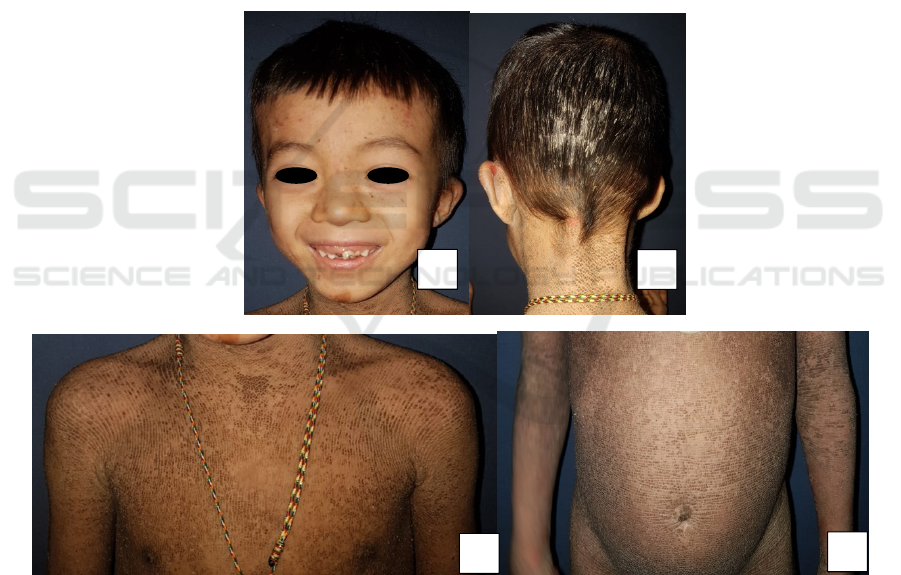
in thick whitish scales.(Figure 1-3).
Figure 1. Clinical manifestation. A. Face is spared B. Whitish thick scales on the scalp C,D. Dirty brown, corrugatedplaques
over the trunk along with areas of superficial erosion.
A
B
C
D
ICTROMI 2019 - The 2nd International Conference on Tropical Medicine and Infectious Disease
258
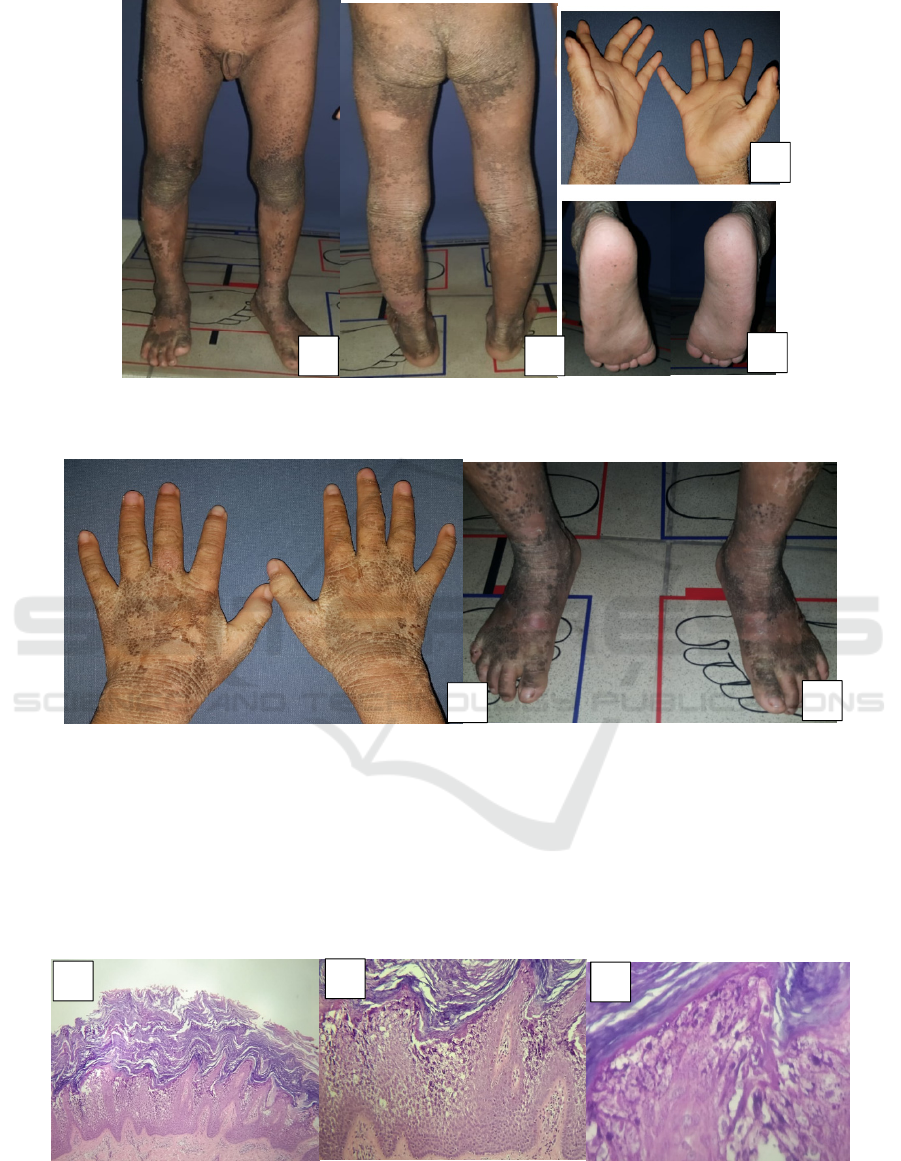
Figure 2. Clinical manifestation. A,B. Hyperkeratotic warty plaques widely spread over the lower limb with areas of
superficial erosion C,D. Sparing palms and soles
Figure 3.Cinical manifestation. A,B. Dirty brown, cobblestone, hyperkeratotic plaques over the posteriorof hand and feet
Histopathology from an area of hyperkeratosis
on his left knee showed massive hyperkeratosis,
acanthosis of stratum malpighi, spongiosis, lysis and
clumping of keratinocytes in the stratum spinosum
to stratum granulosum. Lymphocytic cells are seen
around blood vessels. The findings were consistent
with EHK. (Figure 4 A-C)
On the basis of clinical and histopathological
feature epidermolytic hyperkeratosis was diagnosed.
The patient was treated with vaseline, coconut oil for
scalp, and antiseptic soap. The patient was consulted
to paediatrician, opthalmologist and dentist.
Figure 4.Routine histopathology (haematoxylin-eosin) revealed massive hyperkeratosis, acanthosis, and spongiosis (A,B)
and lysis and clumping of keratinocytes (C)
A
C
B
A B
C
D
A
B
A Rare Case of Epidermolytic Hyperkeratosis: Recognition of Distinctive Clinical and Histopathological Signs
259

3 DISCUSSION
Our case was presented with classical symptoms and
signs of EHK: dirty brown, corrugated
hyperkeratotic plaques distributed all over the body,
with history of trauma-related blistering a few day
after birth that decrease by the age only to be
subsequently replaced by malodorous
hyperkeratosis. This clinical description is
characteristic. Brocq first described EHK in 1902,
and coined the term bullous ichthyosiform
erythroderma, to distinguish it from the non-
blistering condition, congenital ichthyotic
erythroderma. It starts at birth with generalized
erythroderma, blisters, and peeling with even mild
trauma, leading to superficial ulcerations. As the
child grows, the erythroderma and blisters decrease,
and the hyperkeratosis increase. The distinct foul
odor is caused by the bacterial colonization of the
macerated scales.
3
From anamnesis we found that parents were not
affected and so were the rest of the family. He is the
only child of healthy and non-consanguineous
parents. This disease is mostly inherited as an
autosomal dominant trait, albeit 50% of cases result
from spontaneous mutations, and recently an
autosomal recessive inheritance has been reported.
Mutations in keratin 1 and 10 encoding genes,
localized on chromosome 12 and 17, respectively,
are responsible for EHK. Mutations in keratin 1
encoding gene are associated with severe
palmoplantar keratoderma, while mutations of
keratin 10 encoding gene are not.
5
The lack of
palmoplantar involvement in this case suggested that
keratin 10 could be involved.
The histopathologic features of our patient is
consistent with epidermolytic hyperkeratosis with
massive hyperkeratosis, acanthosis, spongiosis, lysis
and clumping of keratinocytes in the stratum
spinsoum to spinosum. The typical histopathologic
features of EHK, which were first described by
Nikolsky in 1897, include acanthosis, marked
hyperkeratosis, coarse keratohyalin granules, and
multiple perinuclear vacuoles present in the upper
spinous layer. Clumping of keratin intermediate
filaments at the suprabasal level can be visualized by
means of electron microscopy, while
immunohistochemistry can show a defect in the
expression of keratin 1 and/or 10.
5
A diagnosis of
EHK is usually made clinically, and can be
confirmed by the presence of typical
histopathological features.
During childhood, EHK can be differentiated
from congenital recessive X-linked ichthyosis on the
basis of the history of blistering and histological
findings. Epidermolytic palmoplanta rkeratoderma is
limited to the palms and soles, whereas ichthyosis
bullosa of Siemens lacks erythroderma, localization
of dark grey hyperkeratosis to the flexural sites, and
areas of peeling of the skin known as the
"Mauserung phenomenon".
5,6
Ichthyosishystrix
Curth-Macklin type patients may look like EHK
patients, but there is no clinical or histological
evidence of blister formation.
5
The patient was treated with vaseline and
coconut oil for thick scales on his scalp, and
antiseptic soap to minimized the foul odor. The
patient was consulted to paediatrician to evaluate the
delayed speech and his general growth and
development, and also to ophtalmologist to evaluate
the involvement of eyes. To evaluate whether he had
dental dysplasia or not, the patient was also
consulted to dentist. Follow up after one month
showed improvement in the hyperkeratotic plaques
and the malodor. Consultations to paediatrician,
opthalmologist and dentist have been done. There
were no abnormalities on his eyes and no dental
dysplasia. The delayed speech was probably caused
by multilingual parents and is not associated with
the disease.
Initial treatment early in disease is targeted
towards symptomatic relief and management of the
secondary complications of the erosions.
5,7,8
These
complications include electrolyte imbalance,
dehydration, infection, and sepsis, especially in
neonates with blisters and erosions. Erosions need to
be managed meticulously with barrier protection and
gentle handling of the skin to minimize the
development of secondary infection. Bacterial
overgrowth, particularly from Staphylococcus
aureus, and an odor can develop as a result of scale
accumulation, which may be controlled with
chlorhexidine or antibacterial cleansers.
5,7,8
Later in
disease, emollients as well as topical and systemic
retinoids can be considered. Topical emollients are
considered mainstay therapy, as well as creams or
ointments that possess keratolytic properties to
reduce the hyperkeratosis scale that develops in
these patients. Examples include urea, alpha-
hydroxy acids, lactic acid, and glycerin, although
lactic acidosis is a concern with topical lactic acid in
infants and small children.
For more severe cases, oral and topical retinoids
have also been shown to improve the skin condition,
although retinoids may promote desquamation and
exacerbate blistering. For unknown reasons,
individuals with keratin 10 gene mutations respond
better to topical or systemic retinoid therapy, as
ICTROMI 2019 - The 2nd International Conference on Tropical Medicine and Infectious Disease
260

compared to those with keratin 1 gene
mutations.
5
Although indicated, we didn’t give
retinoids in this patient due to a lack of availability
in Indonesia. After one month followed up, parents
reported improvement, so we decided to continue
the current therapies.
Education is important in this case. After genetic
counseling, we gave the parents and family
information about high protein diet, patient higiene
in order to prevent secondary infection and
minimized malodor, and also how to keep the patient
from overheating because in patients with
ichthyosis, sweating is often inadequate owing to the
occlusion of eccrine ducts. Affected individuals
should be guarded against overheating during winter
months and kept in air-conditioning during warmer
months, with frequent wetting of the skin or even
cooling suits during sports activities.
9
For the prognosis, widespread blistering clears after
newborn period while the hyperkeratotic scale
usually lifelong. Generalized involvement may
improve to localized disease after puberty.
4 CONCLUSION
Epidermolytic hyperkeratosis is rare and has a
challenging differential diagnosis. Nevertheless, it is
important for clinicians to identify the disease early
in order to reduce morbidity and mortality.
REFERENCES
Fleckman P, DiGiovanna JJ, Elder JT. The Ichthyoses. In:
Goldsmith LA, Katz SI, Gilchrest BA, Paller AS,
Leffell DJ, Wolff K, Eds. Fitzpatrick's Dermatology in
General Medicine. 8
th
Ed. New York: McGrawHill;
2012. p.507-38.
Spitz, Joel L.Genodermatoses: A Clinical Guide to
Genetic Skin Disorders. 2
nd
Ed. Philadelphia:
Lippincott Williams & Wilkins; 2005. p. 6-9.
Hayashida MT, Mitsui GL, Reis NI, Fantinato G, Neto
DJ, Mercante AMC. Epidermolytic Hyperkeratosis -
case report. An Bras Dermatol. 2015;90(6):888-91.
Pan K, Chakrabarti S, Choudhury R, Sarkar A, Mitra
R.Epidermolytic hyperkeratosis: a rare case. Int J Res
Med Sci. 2014 Feb;2(1):347-9.
Alshami M, Mohana M, Alshami A. Epidermolytic
Hyperkeratosis: A Case Report from Yemen. Invest
DermatolVenerol Res. 2016;2(1):61-3.
Charlene U, Ang-Tiu, Marie Eleanore O,
Nicolas.Ichthyosis bullosa of Siemens. J Dermatol
Case Rep. 2012; 6(3):78-81.
Siavash M, Shanehsaz, Bittar R, Anis A, Ishkhanian S.
Bullous ichthyosiformerythroderma: case presentation.
Journal of Pakistan Association of Dermatologists.
2014;24 (1):86-8.
Millsop JW, Sivamani RK, Lee DC, Konia T,
Subramanian N and Fung MA.A Case of Bullous
Congenital IchthyosiformErythroderma, a Rare
Pediatric Genodermatosis, in a Newborn. Austin J
Dermatolog. 2014;1(4):1016.
Paller A, Mancini AJ. Hurwitz clinical pediatric
dermatology: A textbook of skin disorders of
childhood and adolescence. 5
th
ed. New York:
Elsevier/Saunders; 2016.
A Rare Case of Epidermolytic Hyperkeratosis: Recognition of Distinctive Clinical and Histopathological Signs
261
