
Magnetic Properties of Edge Hydrogenated Zigzag Black
Phosphorene Nanoribbon by Fe Doping
Xinfeng Li
1
, Kai Zhang
1
, Shanling Ren
2
, Yunhui Wang
2
and Zhihong Yang
2*
1
College of Electronic Science and Optical Engineering, Nanjing University of Posts and Telecommunications, Nanjing,
China
2
Faculty of Science, Nanjing University of Posts and Telecommunications, Nanjing, China
Keywords: two-dimensional material, black phosphorene nanoribbon, magnetic, first-principles calculation
Abstract: Using first-principles based on density functional theory within the GGA+U framework, we investigated the
structural states, energy, and magnetic properties of edge hydrogenated Zigzag black Phosphorene
Nanoribbon (H-ZPNR) with P atom substituted by Fe atom in different site. Undoped Edge hydrogenated
ZPNR has no magnetic moment, however our results show that the magnetic moment of doped systems
varied from 1μB to 4μB, and the coupling between the Fe atom and adjacent P atoms is either ferromagnetic
or anti-ferromagnetic.
1 INTRODUCTION
Two-dimensional(2D) material displayed great
potential applications in optoelectronic devices,
spintronic, lithium batteries and gas sensors (Liu et
al., 2017; Sun et al., 2016). Single layer black
phosphorus is a two-dimensional structure followed
by graphene and transition metal dichalcogenides
(MoS
2
), stanene and germanene, which had attracted
extensive research efforts. Many two-dimensional
materials are composed of nonmagnetic atoms, and
no magnetic state appeared in the 2D system.
Studies has shown that magnetic properties of
two-dimensional materials can be induced by doping,
vacancy defects, and edge effects (Cao et al., 2018;
Sharma et al., 2018; Son, 2006). For example, magnetic
properties are induced by doping transition metal
atom for MoS2, germanene, and graphene.
Phosphorene monolayer exhibits pleated structure
and is semiconductor with a direct band gap of
0.91eV
(Luan et al., 2017). The field effect transistor
fabricated with phosphorene exhibits a high-speed
carrier mobility (~1000cm
2
V
-1
s
-1
) and gives a high
switching ratio of ~10
5
at room temperature (Li et al.,
2014), indicating the great potential application to
micro-electronic devices. So we explore the physical
properties of edge hydrogenated zigzag black
phosphorene nanoribbon (H-ZPNR) using
first-principles calculation. Latest studies indicated
H-ZPNR does not show edge magnetism and the
system is a direct band gap semiconductor (Zhou et
al., 2017). Moreover, due to the quantum
confinement effect, the band gap of H-ZPNR varies
greatly with the width of the nanoribbon (Peng et al.,
2014). Compare with other transitional atom Co and
Ni, the Fe atom has smaller mass so it can be easily
cooperated into ZPNR and the magnetic
configuration is simple compared with the Mn atom.
In this paper, we explore the electronic structure for
H-ZPNR doped with Fe atom, especially in
magnetic properties.
2 CALCULATION MODEL AND
PARAMETERS
The bare edge zigzag black phosphorus nanoribbon
(ZPNR) show either semiconductor or metallic
behavior in dependence on their edge chemical
groups (Peng et al., 2014), and edge hydrogenated
ZPNR transformed into a direct band gap
semiconductor (Zhu et al., 2014). H-ZPNR are more
stable than the ZPNR, and it is found that the ZPNR
band gap decreases with the increase of the
nanoribbon width (Zhu et al., 2014), we consider the
effect of inequivalent dopant position from the
centre of the nanoribbon to the margin, so we
choose 8 as the width of ZPNR (H-8ZPNR) as
228
Li, X., Zhang, K., Ren, S., Wang, Y. and Yang, Z.
Magnetic Proper ties of Edge Hydrogenated Zigzag Black Phosphorene Nanoribbon by Fe Doping.
DOI: 10.5220/0008188002280231
In The Second International Conference on Materials Chemistry and Environmental Protection (MEEP 2018), pages 228-231
ISBN: 978-989-758-360-5
Copyright
c
2019 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
narrow as possible. We have employed the method
of projector augmented wave based on density
functional theory as implemented in the Vienna
Ab-initio Simulation Package (VASP) (Kresse and
Furthmüller, 1996), and selected the projection
augmented wave (PAW) to describe the mutual
interaction between the ions. Meanwhile, we use the
generalized Gradient Approximation (GGA) of the
Perdew Burke Ernzerhof (PBE) functional form to
process the exchange-correlation energy between
electrons (Perdew et al., 1996a; Perdew et al.,
1996b). Considering that the localized d orbital
electrons will have a significant electron correlation
effect on the transition metal magnetism, correction
method GGA+U is applied to the doping system
(Dudarev et al., 1998). U=2.5eV is used as the
available value for obtaining the magnetic moment
of the transition metal atom in the study of transition
metal doped in single layer black phosphorus (Zhai
et al., 2017). We selected GGA+U (U=2.5eV)
framework to check the magnetic states of doped
systems.
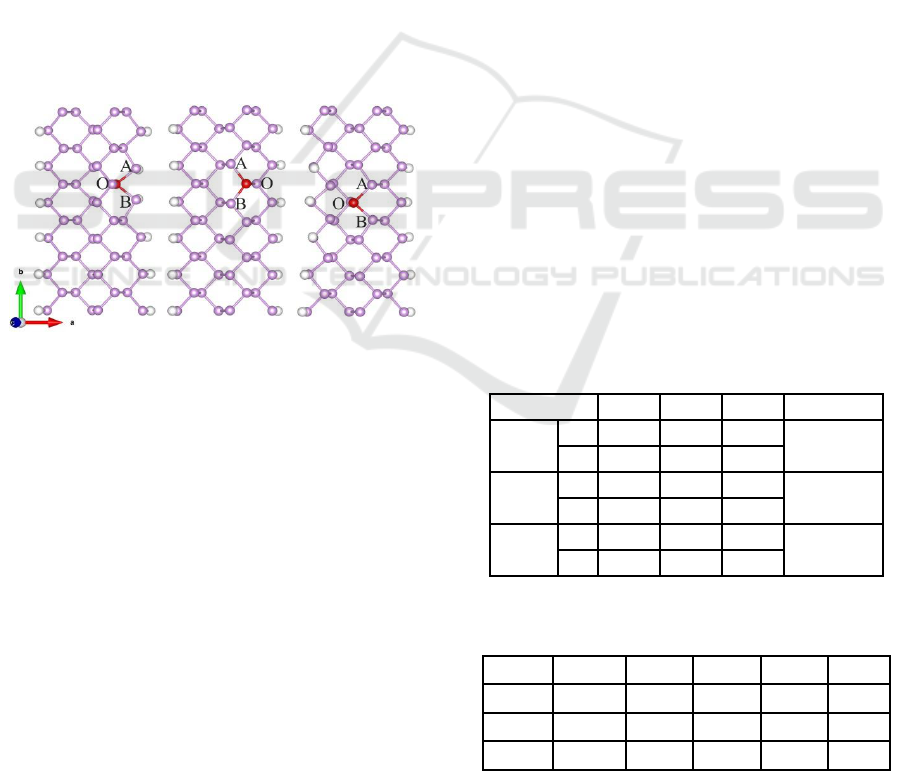
(a)ZFe01 (b)ZFe02 (c)ZFe03
Figure 1: Schematic illustration: (a) ZFe01, (b) ZFe02, (c)
ZFe03. The Fe , P and H atoms are represented by the red,
purple and white color. O, B and A represent P atoms
adjacent to Fe atom.
As shown in Figure 1, the width is defined by the
number of P atoms along the x-axis. We construct
H-8ZPNR with periodic boundary, the vacuum
region of 15Å along the z-axis to avoid the influence
of interaction between the periodic nanoribbons.
And the 6Å vacuum region along the edge of the
hydrogenated nanoribbon to avoid interaction
between H atoms, lattice constant of a = 15.00 Å, b
= 19.80 Å and c = 15.0 Å are used. H-8ZPNR
contains 48 P atoms, the doping concentration of Fe
atom is 2.83%. These inequivalent doping sites were
chosen to research the effect on nanoribbion, three
doped systems substituted Fe atom for original P
atom are defined ZFe01, ZFe02 and ZFe03 from
margin to centre of nanoribbon respectively. The
kinetic energy cutoff of 450eV for the plane wave
expansion, and the structure relaxation was executed
until force on each atom less then 0.01 eV/Å, and
the energy convergence criterion of the each
electron was kept 10
-5
eV. The spin is considered in
the calculation performance. The Monkhorst-Pack
k-point sampling with a 1×7×1 k-mesh is used in the
Brillouin zone integration. And 1×31×1 k-mesh is
used for density of the state.
3 CALCULATION MODEL
Structural States and Energy. The top view of the
ZFe01, ZFe02 and ZFe03 are listed in Figure 1, P-P
bond transforms into Fe-P bond after doping. Three
P atoms adjacent to Fe atom are denoted as ‘O’, ‘A’
and ‘B’. Table 1 shows bond length and energy of
doped system, focus on position ‘O’ in ZFe01, the
P-P(O) bond length is 2.255 Å, the bond length of
Fe-P(O) is 2.222 Å after doping, the difference is
0.033 Å, the difference in ‘B’ and ‘A’ are 0.042 Å
and 0.041Å respectively. The total bond length
difference is 0.116 Å, 0.105 Å and 0.129 Å for
ZFe01 ZFe02 and ZFe03, and ZFe02 has the
smallest change of bond length indicating that the
bond length is likely to be impacted by doping
position. By comparing the energy difference of the
system, ZFe03 has the lowest energy of -299.280eV.
Results indicated that ZFe03 is the most stable.
Table 1: Optimized Bond Lengths ((P-P and Fe-P)in Å),
Energy (in eV).
P(O)
P(B)
P(A)
Energy
ZFe01
P
2.255
2.259
2.26
-297.225
Fe
2.222
2.301
2.301
ZFe02
P
2.255
2.224
2.224
-296.737
Fe
2.311
2.247
2.25
ZFe03
P
2.25
2.224
2.224
-299.280
Fe
2.26
2.28
2.287
Table 2: Magnetic moment of the Fe doped system
under GGA+U(in μB).
Fe
P(O)
P(B)
P(A)
total
ZFe01
2.701
0.09
-0.077
-0.077
2.781
ZFe02
3.426
0.109
0.12
0.118
4.034
ZFe03
1.154
-0.028
-0.042
-0.026
1.07
Magnetic Properties of Edge Hydrogenated Zigzag Black Phosphorene Nanoribbon by Fe Doping
229
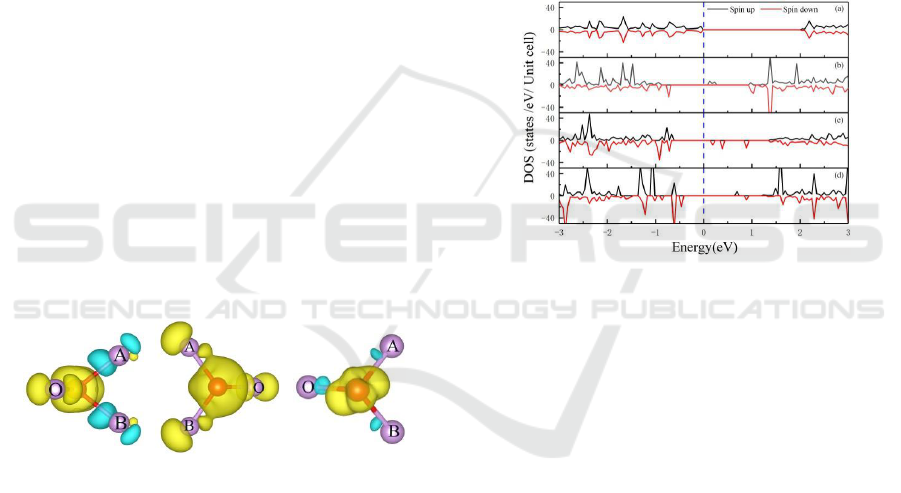
Magnetic Properties: Charge Analysis and
Density of States. The total magnetic moments of
ZFe01, ZFe02 and ZFe03 are 2.78μB, 4.034μB and
1.070μB respectively, ZFe02 has the largest total
magnetic moment, as shown in Table 2. The
magnetic moments of the three doped systems are
mainly arised from the Fe atom. The three P atoms
adjacent to the Fe atom in ZFe02 exhibit spin-up
magnetic moment, indicates that the three adjacent P
atoms are ferromagnetic coupling. In contrast, the
three P atoms adjacent to the Fe atom in the ZFe01
and ZFe03 exhibit spin-down status as a whole in
Figure 2, indicating antiferromagnetic coupling
between Fe atom and adjacent P atoms. The average
magnetic moment of adjacent P atoms and second
nearest P atom’s are 0.081μB and 0.013μB for
ZFe01, 0.116μB and 0.018μB for ZFe02, and
0.032μB, 0.013μB for ZFe03. So the most magnetic
moment are mainly from the Fe and adjacent P
atoms and we can take the magnetic moment of
doped system are localized. The magnetic moment
is relavent to the bond length, with the shorter Fe-P
bond length in ZFe02, the wave function overlapped
more compared with the others, so the magnetic
moment of ZFe02 is larger than others. Especially
the coupling between the Fe atom and the adjacent P
atoms can be either ferromagnetic or
antiferromagnetic indicates the coupling is intricate,
which need further research like multi-dopant
investigation.
(a)ZFe01 (b)ZFe02 (c)ZFe03
Figure 2: Spin charge density for Fe-doped system within
the GGA+U framework. The gold and cyan color have
iso-values of ±0.005 e/A
3
.
The Bader charge analysis has used to
investigate properties of the magnetic moment
between Fe and adjacent P atoms. The localized spin
charge density of the Fe atoms and three adjacent P
atoms were listed, gold color represents the spin-up
density, and cyan color represents the spin-down
density. In ZFe01 and ZFe03, the gold areas are
mainly accumulated around the Fe atom, the cyan
areas are small and concentrated in P atoms adjacent
to the Fe atom. Therefore, the adjacent P atoms of
Fe atoms in ZFe01 and ZFe03 are antiferromagnetic
coupling. However, ferromagnetic coupling is
exhibited in the ZFe02, which is consistent with the
conclusion from magnetic moment analysis. Thus
the spin charge density of the system is mainly
concentrated around the Fe atom, there are only a
few spin charge densities around the adjacent P
atoms, and there are almost no spin charge densities
in the adjacent position and locations far away from
the Fe atom. It also shows that the magnetic
properties in the doped system are mainly from the
Fe atom. Bader charge analysis also illustrate the
charge transfer, the Fe atom in three doped systems
are all electronegative, and electrons transfer from
the d orbital of the Fe atom to the p orbital of the P
atoms, and the charge transfer electrons are 0.37e,
0.44e, and 0.33e, respectively.
Figure 3: DOS illustration: (a) ZPNR, (b) ZFe01, (c)
ZFe02, (d) ZFe03.
Density of states (DOS) for ZFe01, ZFe02 and
ZFe03 are listed in Figure 3. In H-8ZPNR, the band
gap is 2eV. However, dopant energy state appeared
in ZFe01, ZFe02 and ZFe03 so reduce the band gap,
their band gap is 0.76eV, 0.74eV and 1.04eV
respectively. The majority spin density of state and
the minority spin density of state are asymmetric,
Compared with the width of band gap, central doped
is greater than the edge doped. As shown in Figure 4,
the partial wave hybridization between d orbital of
the Fe atom and the p orbital of P atoms make most
contribution to density of states (PDOS) clearly
indicate the influence of electron orbital of the
atom on the magnetic properties. The magnetic
moment is derived from d orbital of the Fe atom, p-d
orbital the total magnetic moment and this coupling
is localized.
MEEP 2018 - The Second International Conference on Materials Chemistry and Environmental Protection
230
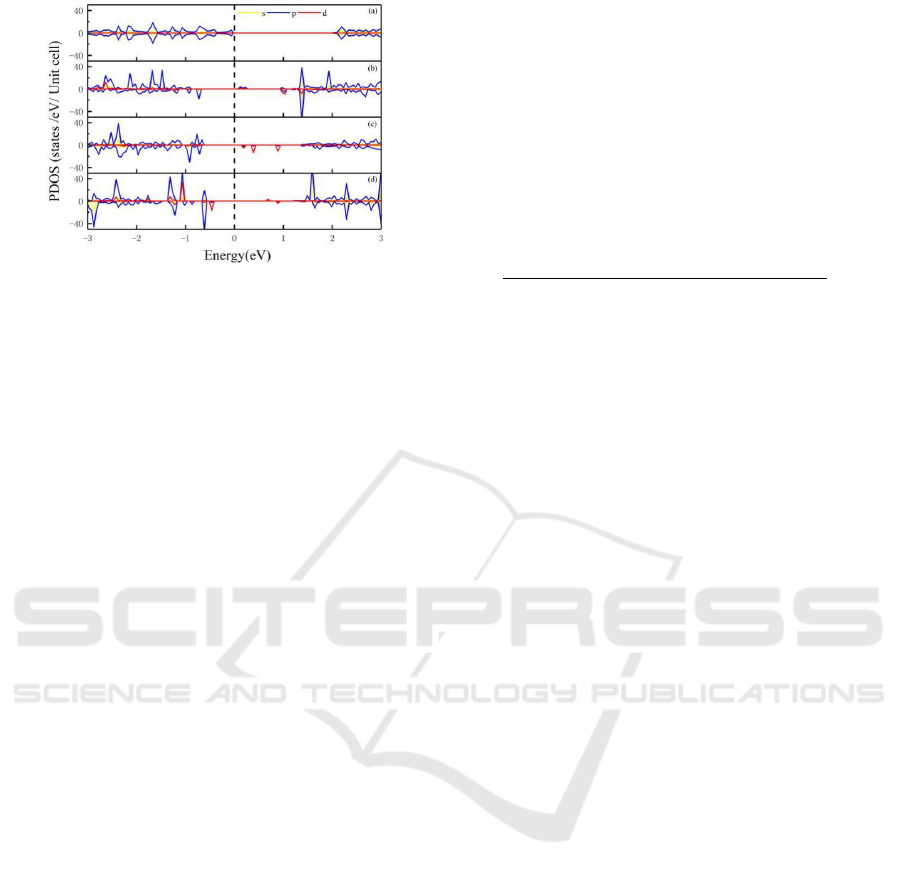
Figure 4: PDOS illustration: (a) ZPNR, (b) ZFe01, (c)
ZFe02, (d) ZFe03.
4 CONCLUSIONS
In summary, based on the density functional
calculations, we have explored the structure and
magnetic properties of doped black phosphorene
nanoribbon. The result show that doped nanoribbon
had lattice distortion but the structure is stable. The
substitution of Fe for P induce magnetic moment
and this moment is localized. The coupling between
the Fe atoms and adjacent P atoms can be either
ferromagnetic or anti-ferromagnetic coupling,
relavent to the magnetic value of Fe atom, which is
required further research.
ACKNOWLEDGEMENTS
This research was supported by Basic Science
Research Program through the Natural Science
Foundation of China (No. 11804169 and 11747029)
and by the Research initiation funds of Nanjing
University of Posts and Telecommunications No.
NY216029. In addition, I appreciate the help from
Dr. Huang Xin for useful discussion.
REFERENCES
Cao, C. et al., 2018. “Transition metal adatom and dimer
adsorbed on graphene: Induced magnetization and
electronic structures”, Physical Review B, Vol. 81 No.
20, pp. 2498-2502.
Dudarev, S. L. et al., 1998. “Electron-Energy-Loss
Spectra and the Structural Stabilityof Nickel Oxide:
An LSDA+U Study”, Physical Review B, Vol. 57 No.
3, pp. 1505-1509.
Kresse, G. and Furthmüller, J., 1996. “Efficient iterative
schemes for ab initio total-energy calculations using a
plane-wave basis set”, Physical Review B, Vol. 54 No.
16, pp. 11169-11186.
Li, L. et al., 2014. “Black phosphorus field-effect
transistors”, Nature Nanotechnology, Vol. 9 No. 5, pp.
372-377.
Liu, S. X. et al., 2017. “Graphene/phosphorene nano-
heterojunction: facile synthesis, nonlinear optics, and
ultrafast photonics applications with enhanced
performance”, Photonics Research, Vol. 5 No. 6, pp.
662- 668.
Luan, Z. et al., 2017. “First-principles study on electronic
structures and magnetic properties of Eudoped
phosphorene”, Superlattices and Microstructures.
http://dx.doi.org/10.1016/j.spmi.2017.07.039
Peng,X. et al., 2014. “Edge effects on the electronic
properties of phosphorene nanoribbons”, Journal of
Applied Physics, Vol. 116 No. 14, p. 144301
Perdew, J. P. et al., 1996a. “Generalized Gradient
Approximation Made Simple”, Physical Review
Letters, Vol. 77 No. 18, pp. 3865-3868.
Perdew, J. P. et al., 1996b. “Generalized Gradient
Approximation Made Simple”. Physical Review
Letters, Vol. 78 No. 7, p. 3865.
Sharma, D. K. et al., 2018. “Magnetism by embedding 3d
transition metal atoms into germanene”, Journal of
Physics D-Applied Physics, Vol. 51 No. 22.
Son, Y. W. et al., 2006. “Half-metallic graphene
nanoribbons”, Nature, Vol 444 No. 7117, pp. 347-349.
Sun, M. L. et al., 2016. “Magnetism in transition metal-
substituted germanane: A search for room temperature
spintronic devices”, Journal of Applied Physics, Vol.
119 No. 14, pp. 666-672.
Zhai, C. Y. et al., 2017. “Strain tuning of magnetism in
transition-metal atom doped phosphorene”,
Superlattices and Microstructures, Vol. 101, pp. 49-
56.
Zhou, W. Z. et al., 2017. “Doping effects on the electronic
properties of armchair phosphorene nanoribbons: A
first-principles study”, Physica E: Low-dimensional
Systems and Nanostructures, Vol. 94, pp. 53-58.
Zhu, Z. L. et al., 2014. “Magnetism of zigzag edge
phosphorene nanoribbons”, Applied Physics Letters,
Vol. 10, p. 113105.
Magnetic Properties of Edge Hydrogenated Zigzag Black Phosphorene Nanoribbon by Fe Doping
231
