
A Case of Majocchi Purpura: Clinical and Histopathological
Approaches for Diagnosis
Fadhli Aulia Mughni, Sarah Mahri, Teffy Nuary, Rahadi Rihatmadja, Kusmarinah Bramono
Department of Dermatology and Venereology, Faculty of Medicine University of Indonesia / Dr. Cipto Mangunkusumo
National General Hospital, Jakarta, Indonesia
Keywords: clinical patterns, diagnosis, histopathology, Majocchi purpura
Abstract: Majocchi Purpura is one of the morphological varieties of pigmented purpuric dermatoses. They are rare
disorders with unknown etiology, and characteristically presents as nonblanching petechiae, pigmentation,
and the presence or absence of telangiectasia with a predilection for the lower extremities. They are usually
asymptomatic and self-limiting, but tend to have a chronic course and are highly recurrent. The diagnostic
approach for patients suspected with pigmented purpuric dermatoses is quite straightforward, with clinical
observation and histopathological examination as the mainstay diagnostic modalities. However, this
disorder may initially show similar manifestations as other cutaneous diseases, such as contact dermatitis,
vasculitis, or mycosis fungoides, making it oftenly misdiagnosed and mistreated by dermatologists. We
present a case of a 28-year-old woman initially referred with polyarteritis nodosum. However, the lesions
were painless and consisted of several hyperpigmented annular patches that clinically resembled Majocchi
Purpura. We submitted the case for biopsy with vasculitis as the differential diagnosis. Histopathological
findings were consistent with pigmented purpuric dermatosis, that we commenced the treatment with
symptomatic measures supplemented with antioxidants. Recognizing distinctive clinical patterns of disease
was indispensable. Laboratory examinations had limitations, and their results needed to be interpreted
within the clinical context. The correct diagnosis will prevent overtreatment and unnecessary healthcare
visit by patient.
1 INTRODUCTION
Pigmented Purpuric Dermatoses (PPD) is a group of
disorders characterized by nonblanching purpuric
rash, leaving residual hyperpigmented patches
mainly on lower extremities.(Sardana, 2004) The
etiology is unknown, although several drugs and
other conditions have been documented. (Devere,
2012) Most patients are asymptomatic or presenting
with mild symptoms. PPD is usually chronic, highly
recurrent, and difficult to treat, although self-
limiting cases have been reported.(Devere, 2012)
This disorder is morphologically categorized into:
(1) Schamberg’s disease (SD), (2) Purpura annularis
telangiectodes of Majocchi (Majocchi Purpura), (3)
Pigmented purpuric lichenoid dermatosis of
Gougerot and Blum (PPLD), (4) Eczematid-like
purpura of Doucas and Kapetanakis, (5) Itching
purpura of Lowenthal, and (6) Lichen aureus
(LA).(Kim, 2015)
PPD is considered uncommon, despite lack of
sufficient data in Indonesia. A
clinicoepidemiological study by Sharma and
GuptaSharma, 2012 in 2012 found 0.18% PPD cases
from 55,323 patients. All races may be affected,
with more occurrence seen in males except in
Majocchi purpura. (Sardana, 2004,Hoesly, 2009)
The majority have history of routine activity that put
constant pressure to the limbs, such as prolonged
standing during work, high-intensity sport, or
repetitive tasks. Several factors is believed to
contribute to its pathogenesis, including capillary
fragility, humoral immunity, cell-mediated
immunity, gravitational forces, venous hypertension,
focal infection, and contact allergy. (Devere,
2012,Sharma, 2012)
Most cases were diagnosed clinically. The
presentations of PPD are usually characteristic, with
pigmented patches and petechiae as the most
consistent findings, without palpable purpura.
However, initial manifestations may easily be
interpreted as other cutaneous diseases, mainly
402
Mughni, F., Mahri, S., Nuary, T., Rihatmadja, R. and Bramono, K.
A Case of Majocchi Purpura: Clinical and Histopathological Approaches for Diagnosis.
DOI: 10.5220/0008158404020405
In Proceedings of the 23rd Regional Conference of Dermatology (RCD 2018), pages 402-405
ISBN: 978-989-758-494-7
Copyright
c
2021 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

because their rare occurrence entices clinicians to
weigh other more common disorders showing
similar features. Pigmented purpuric dermatoses
need to be distinguished from contact dermatitis,
stasis dermatitis, angioma serpiginosum, mycosis
fungoides, and most importantly, vasculitis.
(Sardana, 2004,Devere, 2012) Histopathology
examination most commonly shows perivascular
infiltrate of lymphocytes around superficial blood
vessels, endothelial cell swelling, erythrocyte
extravasation, and hemosiderin deposition. (Sardana,
2004-Kim, 2015) While crucial for confirming the
clinical diagnosis of PPD, skin biopsy is also
important to exclude cutaneous T-cell lymphoma,
which in its early stages closely resembles PPD.
(Sardana, 2004)
No satisfying therapy has been found for PPD.
Some lesions have been reported to subside
spontaneously, however numerous agents have been
used to alleviate the symptoms and skin lesions,
including topical corticosteroids, antihistamines,
bioflavonoids, ascorbic acid, griseofulvin,
pentoxifylline, cyclosporine, and phototherapy, with
variable but inconclusive outcomes. (Sardana, 2004)
Misdiagnosis may lead to overtreatment and
unnecessary healthcare visits, therefore it is crucial
for dermatologists to be able to recognize PPD
lesions and manage the patients accordingly.
2 CASE
A 28-year-old woman was referred to the
Dermatovenerology clinic, Cipto Mangunkusumo
National Central General Hospital with the diagnosis
of polyarteritis nodosum. She presented with
multiple brownish-red lesions on the legs that have
been present for four months. Initially,
asymptomatic red spots appeared on both feet. They
spread upward reaching up to the thighs.
Approximately two weeks later, some lesions faded
to brownish discoloration. The lesions were more
noticeable in relatively cold environment, such as in
air-conditioned room. She also occasionally
complained of ankle joints pain since two months
before presentation. She took oral B-complex
vitamins, but to no avail.
Physical examination was unremarkable except
the presence of multiple brownish-purpuric patches
bilaterally on the arms, lower legs, and feet. Some of
the macules were annular and reticular. No palpable
purpura was observed. (Figure 1).
Figure 1. Multiple petechial and hyperpigmented
patches in annular (arrow) and reticular (circle)
configurations.
Past medical history was unremarkable. Previous
use of medications was denied. The patient usually
wore high-heeled shoes for work, which require
repeated walking between offices during working
hours. Initial laboratory examinations showed mild
leukopenia, eosinophilia, and high erythrocyte
sedimentation rate. The results of hemostasis,
urinalysis, as well as blood chemistry examinations
were within normal ranges. At the Internal Medicine
clinic, she was also examined for anti-nuclear
antibodies (ANA), Hepatitis B surface antigen
(HbsAg), anti-hepatitis C virus (anti-HCV), humman
immunodeficiency virus (HIV), mixed activated
partial thromboplastin time (APTT), and lupus
anticoagulant to rule out other diagnostic
possibilities. All those results were negative, except
for ANA.
We diagnosed this patient with Majocchi
purpura. Schamberg disease, another type of PPD,
and vasculitis was also considered. The patient was
then sent for biopsy. Histopathology from two
different sites revealed superficial perivascular
lymphocytic infiltrates, endothelial cell swelling,
and erythrocytes extravasation, which are consistent
with PPD. (Kim, 2015) (Figure 2)
The patient has been followed up for two
months, and was treated symptomatically with
emollients and antioxidants, including ascorbic acid
500 mg daily. Topical corticosteroid twice daily was
started one month ago because the patient exhibited
mild occasional pruritus. There was slight
improvement of the patient’s skin lesions, without
worsening of symptoms or appearance of new
purpuric patches
.
A Case of Majocchi Purpura: Clinical and Histopathological Approaches for Diagnosis
403
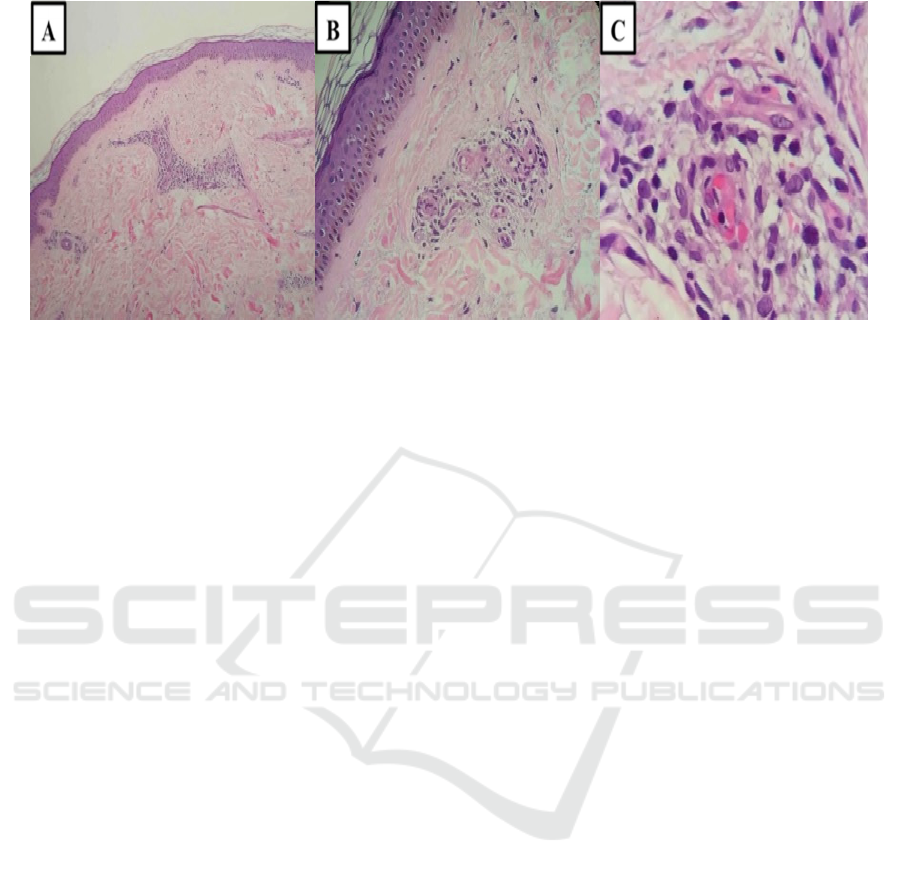
Figure 2. (A) Perivascular lymphocytic infiltrates in the upper dermis (H&E, 50X); (B) Endothelial cell swelling,
superficial perivascular lymphocytes (H&E, 100X); (C) Erythrocytes extravasation around the superficial blood vessel
(H&E 400X).
3 DISCUSSION
The clinical appearance of pigmented purpuric
dermatoses may be similar to cutaneous vasculitis.
However, the purpuric patches in cutaneous
vasculitis are palpable, unlike in PPD. (Devere,
2012) Our patient was initially assessed as
polyarteritis nodosa (PAN), a necrotizing vasculitis
of medium-sized vessels that commonly affected the
nerves, intestinal tract, heart, and joints. Indeed, the
initial manifestations of some cases of PAN are skin
lesions. (Kazandjieva, 2017) However, she did not
complained of constitutional symptoms, such as
fever, malaise, weight loss, or abdominal pain, and
did not exhibit characteristic subcutaneous nodules.
Therefore, it was important to rule out related
systemic diseases, since the patient also complained
of ankle pain. There were several case reports
associating persistent PPD with CTCL and mycosis
fungoides, which can all be excluded by
histopathologic examination. (Toro, 1997,Martinez,
2001)
The reticular pattern of the macules and
exacerbation by cold suggested the possibilities of
disturbance of blood flow to the skin in response to
low temperature exposure, such as seen in livedo
reticularis. Occlusions of vessels in livedo reticularis
may be found in vasculopathy disorders, such as
antiphospholipid syndrome (APS) and
cryoglobulinemia. (Gibbs, 2005) The negative
results of HBsAg, anti-HCV, HIV, mixed APTT,
and lupus anticoagulant examinations of this patient
ruled out those diseases. Anti-nuclear antibodies
were positive, however it does not entirely confirm
the presence of an autoimmune disease. (Pisetsky,
2011) Our patient’s history and clinical appearance
does not support an autoimmune disease, but
continuous observation is needed.
Typical histopathologic findings of PPD
mentioned in literatures are perivascular infiltrate of
lymphocytes in superficial dermis, endothelial cell
swelling, erythrocytes extravasation, and
hemosiderin deposition. (Devere, 2012,Kim, 2015)
Superficial perivascular lymphocytes were clearly
observed in our patient, as well as endothelial
swelling and extravasation of erythrocytes in the
dermis. However, in two biopsy specimens we could
not find deposition of hemosiderin. Although it is an
important feature for distinguishing PPD from other
disorder, hemosiderin deposition is not always
found. In a retrograde analysis of 113 patients with
PPD in Korea, hemosiderin deposition was found in
only 26,3% subjects, while perivascular lymphocyte
infiltration and erythrocyte extravasation were found
in 79% and 50% patients, respectively.(Kim, 2015)
It is advisable to do histochemical staining with
Perls stain to identify hemosiderin more easily, as
well as with Masson-Fontana stain to exclude
melanin pigment. (Kazandjieva, 2017) Focal
karyorrhectic nuclear dust may occasionally be
found, especially in active pronounced lesions,
alongside with narrowing of vessel lumens.
(Kazandjieva, 2017,Weedon, 2010) Both features
were found in this patient.
The patient was treated with antioxidants and
symptomatic therapies, which consist of emollient,
anti-inflammatory and antipruritic agents. Treatment
of PPD is difficult, and results in literatures are
inconsistent. (Hoesly, 2009) proposed a systematic
approach for the management of Majocchi Purpura.
Through review of available literatures, the scheme
RCD 2018 - The 23rd Regional Conference of Dermatology 2018
404

suggested treatment of this disorder according to the
symptoms severity and use of combination treatment
if needed.
4 CONCLUSION
We reported a case of Majocchi purpura in a patient
who was initially assessed as a polyarteritis
nodosum. Although uncommon, it is important for
dermatologists to be able to recognize the
characteristic PPD lesions, and perform the
necessary examinations to rule out other suspected
disorders. Histopathology examination is until now
the most important tool to confirm the diagnosis of
PPD. Pigmented purpuric dermatoses is highly
recurrent, although some lesions are self-limiting.
Treatment is seldom satisfactory, and remains a
challenge for clinicians.
REFERENCES
Devere, T.S., Patel, A.B., 2012. Pigmented purpuric
dermatoses. In: Goldsmith LA, Katz SI, Gilchrest BA,
Paller AS, Leffel DJ, Wolff K, editors. Fitzpatrick’s
Dermatology in General Medicine. Vol 1. 8th ed. New
York: McGraw-Hill, p. 2049-2051.
Gibbs, M. B., English III, J. C., & Zirwas, M. J., 2005.
Livedo reticularis: an update. Journal of the American
Academy of Dermatology, 52(6), pp. 1009-1019.
Hoesly, F. J., Huerter, C. J., & Shehan, J. M., 2009.
Purpura annularis telangiectodes of Majocchi: case
report and review of the literature. International
journal of dermatology, 48(10), pp. 1129-1133.
Kazandjieva, J., Antonov, D., Kamarashev, J., & Tsankov,
N., 2017. Acrally distributed dermatoses: vascular
dermatoses (purpura and vasculitis). Clinics in
dermatology, 35(1), pp. 68-80.
Kim, D. H., Seo, S. H., Ahn, H. H., Kye, Y. C., & Choi, J.
E., 2015. Characteristics and clinical manifestations of
pigmented purpuric dermatosis. Annals of
dermatology, 27(4), 404-410.
Martínez, W., Del Pozo, J., Vázquez, J., Yebra-Pimentel,
M. T., Almagro, M., García-Silva, J., & Fonseca, E.,
2001. Cutaneous T-cell lymphoma presenting as
disseminated, pigmented, purpura-like
eruption. International journal of dermatology, 40(2),
pp. 140-144.
Pisetsky, D. S., 2011. Antinuclear antibodies in healthy
people: the tip of autoimmunity's iceberg?, 13(2), 109.
Sardana, K., Sarkar, R., & Sehgal, V. N., 2004. Pigmented
purpuric dermatoses: an overview. International
journal of dermatology, 43(7), pp. 482-488.
Sharma, L., & Gupta, S., 2012. Clinicoepidemiological
study of pigmented purpuric dermatoses. Indian
dermatology online journal, 3(1), 17, pp. 12-20.
Toro, J. R., Sander, C. A., & LeBoit, P. E., 1997.
Persistent pigmented purpuric dermatitis and mycosis
fungoides: simulant, precursor, or both?: a study by
light microscopy and molecular methods. The
American journal of dermatopathology, 19(2), pp.
108-118.
Weedon, D., 2010. The vasculopathic reaction pattern.
In: Weedon’s skin. Pathology. 3rd ed. Elsevier, MO,
pp. 195.
A Case of Majocchi Purpura: Clinical and Histopathological Approaches for Diagnosis
405
