
Malignant Peripheral Nerve Sheath Tumor in Neurofibromatosis
Type 1 Patient
Sudarsono
1
, Obdes Maharni Emputri
2
, Iswahyudi
3
, Indriasari
4
, Tati Herdawati
5
, Dyah Marianingrum
5
1
Department of Dermatovenereology, Raja Ahmad Tabib General Hospital, Tanjungpinang, Indonesia
2
Department of Ophthalmology, Raja Ahmad Tabib General Hospital, Tanjungpinang, Indonesia
3
Department of Surgery, Raja Ahmad Tabib General Hospital, Tanjungpinang, Indonesia
4
Department of Radiology, Raja Ahmad Tabib General Hospital, Tanjungpinang, Indonesia
5
Department of Pathology, Raja Ahmad Tabib General Hospital, Tanjungpinang, Indonesia
Keywords: neurofibromatosis type 1, malignancy, malignant peripheral nerve sheath tumor
Abstract: Malignant peripheral nerve sheath tumor (MPNST) is rare and highly aggressive neoplasm. Approximately
half of MPNST cases occur in association with neurofibromatosis type 1 (NF1). MPNST contribute
significantly in reducing life-span of neurofibromatosis type 1 patients. The only known definitive therapy
for MPNST is surgical resection with wide negative margins which may not be feasible in certain conditions
involving tumor size, location, and/or metastases. Therefore, early diagnosis of MPNST is mandatory to
increase the rate of successful surgical resection. We present a 36-year-old female with NF1, who had a
giant MPNST on the right arm. She died six months after diagnosis in spite of a surgical resection and
chemotherapy. This case report aimed to highlight the signs and symptoms of malignant peripheral nerve
sheath tumor and examinations that are needed to make an early diagnosis of MPNST especially in patients
with neurofibromatosis type 1.
1 INTRODUCTION
Neurofibromatosis type 1 (NF1) is an autosomal
dominant disorder characterized by neurofibromas,
cafe-au-lait spots, intertriginous freckling, bone
malformations, learning disabilities and iris
hamartomas. It is associated with mutation in NF1, a
tumor suppressor located on chromosome 17q11.2.
NF1 encodes neurofibromin, a protein of the ras-
signal transduction pathway (Zehou et al., 2013).
NF1 represents a major risk factor for development
of malignancy, particularly malignant peripheral
nerve sheath tumors (MPNST), optic gliomas, other
gliomas, and leukemias (Korf, 2005).
MPNST is a rare disease with an incidence of 1
in 1.000.000 in the general population. The
incidence of MPNST in patients with NF1 has been
estimated to be 2% to 5%. MPNST is considered
aggressive and is associated with a low survival rate
(Hwang et al., 2017). MPNST contribute
significantly in reducing life-span of NF1-patients
(Freidrich, 2007). We present a rare case of giant
MPNST with lung metastases in a patient with NF1.
2 CASE
A 36-year-old female presented with painful huge
mass on her right arm, which rapidly increased in
size within a four-year period. The mass had
restricted movement of the extremity without
neurological deficits. She also had multiple nodular
masses of different sizes almost covering her entire
body. These masses were initially seen in the head,
neck and upper extremities when she was 7 years
old. She also had multiple hyperpigmented macules
on her axilla and trunk when she was 3 months old.
None of her close relatives had similar signs and
symptoms.
Upon physical examination, a firm, tender soft
tissue mass with small necrotic area in right arm was
found. Size of the mass was about 13 cm x 9 cm x 9
cm (Figure 1). “Bag of worm” sensation was
negative on palpation. Multiple soft dome-shaped,
flesh-colored nodules with diameters ranging from
0,5 cm to 5 cm covering almost her entire body
(Figure 1). Multiple small to large cafe-au-lait
macules, more than six in number, with irregular
margins were seen on trunk. She also had multiple
Sudarsono, ., Emputri, O., Iswahyudi, ., Indriasari, ., Herdawati, T. and Marianingrum, D.
Malignant Peripheral Nerve Sheath Tumor in Neurofibromatosis Type 1 Patient.
DOI: 10.5220/0008157403590362
In Proceedings of the 23rd Regional Conference of Dermatology (RCD 2018), pages 359-362
ISBN: 978-989-758-494-7
Copyright
c
2021 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
359
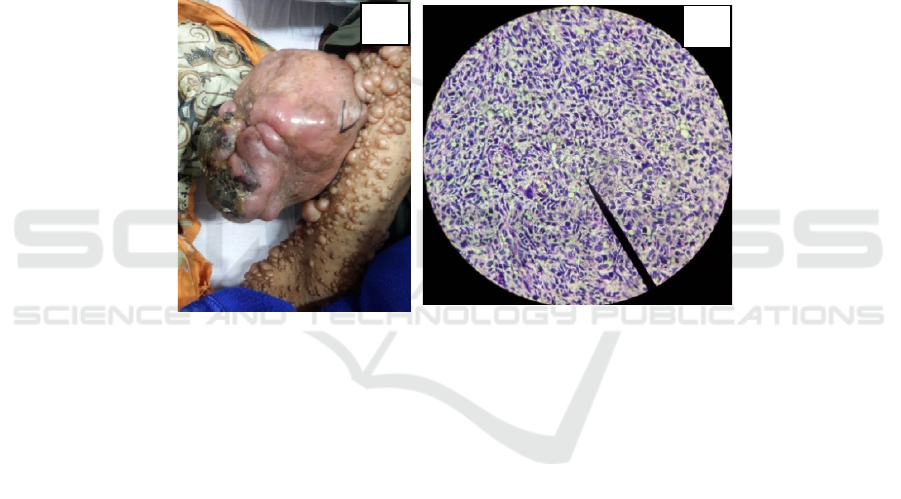
tiny hyperpigmented macules in her both axilla. Eye
examination revealed multiple Lisch nodules on the
iris of both eyes and the visual acuity were 6/12.
Chest x-ray examination was normal.
Based on anamnesis and physical examination,
the differential diagnosis of the enlarging mass were
MPNST and plexiform neurofibroma (PN). The
patient underwent wide excision under general
anesthesia. Histopathology evaluation revealed
overall features of MPNST composed of
pleomorphic spindle cells with hyperchromatic
nuclei and mitotic activity (more than 20 mitoses per
10 high-power field) arranged curlicue and whorls
(Figure 2). Infiltration of the tumor cells into the
surrounding soft tissue and area of necrosis was
noted. Immunohistochemical analysis revealed a
scattered positive staining uptake in the cells with S-
100 antibody.
During follow-up, three months later, she
presented with enlarging mass of 6 cm x 8 cm in her
right axilla. Fine needle aspiration biopsy of the
mass revealed disease progression presence of
mitotic cells dominance, including some
pleomorphic malignant spindle cell proliferation.
CT-scan examination of axilla and lung revealed
solid mass of 6,2 cm x 8,3 cm x 6,1 cm on axilla and
multiple nodules on lung suggestive lung metastases.
We referred the patient to oncology surgeon and she
received two cycles of chemotherapy (ifosfamide)
without any improvement. She died six months after
diagnosis.
Figure 1: (A) A huge soft tissue mass with multiple neurofibromas in right arm. (B) Leomorphic spindle cells with
hyperchromatic nuclei and mitotic activity.
3 DISCUSSION
The term MPNST currently describes a
heterogeneous group of malignant tumors that
possibly arise from cells of the nerve sheath
(Freidrich et al., 2007). MPNST may appear de novo
or develop from the malignant transformation of a
benign neural neoplasm, generally a PN.
Approximately half of MPNST cases occur in
association with NF1 (Cunha et al., 2012).Leroy et
al (2001) reported the prevalence of MPNST was
approximately 4% in patients with NF1.In our
patient, MPNST occurred in association with NF1.
A diagnosis of NF1 was confirmed by the presence
of four clinical manifestations that met the National
Institutes of Health (NIH) consensus criteria.
In NF1 patients, one allele of Nf1 is inactivcated.
PN is initiated when a second-hit mutation
inactivates the remaining functional Nf1 gene,
resulting in a loss of neurofibromin expression and
Ras hyperactivation. This enhanced Ras signalling
promotes the proliferation and invasive behavior of
the neoplastic cells and their production of factors
that recruit other Nf1 happloinsufficient cell types
into the nascent neurofibroma. The subsequent loss
of additional tumor suppressor genes (p53, cyclin
D1-cyclin-dependent kinase (CDKN2A), phosphate
and tensin homologue (PTEN), retinoblastoma
(Rb)), amplification of key growth factor receptor
genes (epidermal growth factor receptor (EGFR),
ErbB2, C-Met, platelet-derived growth factor
receptor (PDGFRα), KIT), mutation of polycomb
repressive complex 2 (PRC2) components and
alteration of key cytoplasmic signaling pathways
then leads to the development of MPNST derived
from the the neoplastic Schwann cells within the
neurofibroma (Carroll, 2016).
MNPST may arise at any age with no gender
predilection (Farid et al., 2014).Patients with NF1
A
B
RCD 2018 - The 23rd Regional Conference of Dermatology 2018
360

usually present at a slightly earlier age than those
with sporadic MPNST (Goldblum et al., 2014). The
median age for sporadic MPNST is between 30 and
60 years, and that for NF1- associated MPNST is
between 20 and 40 years (Farid et al., 2014). Our
case was a 36-year-old female.
MPNST usually presents as a progressively
enlarging mass with pain and, later neurological
symptoms such as weakness and paresthesias.
Symptoms are present for months before the
malignant neoplasm is identified correctly (Farid et
al., 2014; Leroy et al., 2001).Most MPNST arise in
association with major nerve trunks, including the
sciatic nerve, brachial plexus, and sacral plexus.
Consequently, the most common anatomic sites
include the proximal portions of the upper and lower
extremities and the trunk. Few MPNST arise in the
head and neck (Goldblum et al., 2014). de
Vasconcelos et al. (2017) reported the extremities
(58%) as the most common affected sites, followed
by trunk (32%) and head and neck (10%) (de
Vasconcelos et al., 2017). MPNST is usually large,
averaging more than 5 cm in diameter and has a
fleshy, opaque, white-tan surface marked by areas of
secondary hemorrhage and necrosis (Goldblum et
al., 2014).de Vasconcelos et al. (2017) reported the
mean tumor sizes were 15,8±8,2 cm (range, 3-47
cm).
10
Our case presented with painful huge mass
(13 cm x 9 cm x 9 cm) on proximal portion of the
upper extremity without neurological symptoms.
Most MPNST are aggressive with a high
likehood of local recurrence and distant metastases.
The local recurrence rate varies from 40% to 65%
and the metastatic rate from 40% to 68%. They
frequently metastasize to the lung followed by bone,
liver, brain, soft tissue, skin and retroperitoneum.
11
Three months after surgical resection, our patient
developed local recurrence in the axilla and lung
metastases.
Fluorodeoxyglucose positron emission
tomography (PET-CT) is of great value in
monitoring lesions with the potential for malignant
transformation in NF1 (Batista et al., 2015).
Increased uptake was found to be characteristic of
MPNST (Korf, 2005). Due to the frequency and
severity of MPNST associated to NF1, PET-CT scan
could be useful in the following situations: a) when
the plexiform tumor growth is inconsistent with the
child’s growth track; b) in the presence of
neurological deficit, c) changes in tumor texture; and
finally d) when patient reports an inexplicable and
progressive pain (Batista et al., 2015).
Histologic features of MPNST are composed of
monotonous spindle cells arranged in intersecting
fascicles. Pleomorphic variants also exist. At low
power, alternating hyper- and hypocellular areas
may be present, often with hypercellular areas
localized in close proximity to blood vessels.
Compared with benign neurofibromas, MPNST
usually demonstrate a marked increase in tumor
cellularity, pleomorphism, and mitotic activity and
show a more organized cellular growth pattern, with
less extracellular matrix material (Farid et al., 2014).
S-100 protein has been the classic and the most
widely used antigen for documenting nerve sheath
differentiation. Between 50% and 90% of MPNST
express the antigen but usually focally only
(Goldblum et al., 2014).Our patient had
histopathological and immunochemical findings
similar to the earlier findings and diagnosis of
MPNST was made.
Current treatment of MPNST is similar to
treatment of soft tissue sarcomas as a whole and
relies primarily on local control measures. The only
known definitive therapy for MPNST is surgical
resection with wide negative margins, which may
not feasible due to variables such as tumor size,
location, and/or metastases. The role of adjuvant
radiation and chemotherapy is not defined (Kim et
al., 2017). Chemotherapy is usually preferred for
metastatic disease and may be also useful in the
preoperative management in order to decrease the
size in patients with inoperable tumors. Radiation
therapy is recommended for positive microscopic
margins providing local control and delaying the
onset of recurrence (Pourtsidis et al., 2014). Our
patient underwent excision and chemotherapy was
given after local recurrence and distant metastases of
the tumor to the lung.
MPNST are very aggressive tumors and all
current treatments have shown poor results. The
five-year overall survival rate of patients with
MPNST ranges between 34% and 58%, with several
studies suggesting that prognosis in the setting of
NF1 may be worse (Dunn et al., 2013).A meta-
analysis testing the effect of NF1 status on MPNST
by Kolberg et al. (2012) showed a significantly
worse outcome in NF1 patients (Kolberg et al.,
2013). de Vasconcelos et al. (2017) showed the
presence of NF1 and tumor size (greater than 10 cm)
had a significant negative impact on overall survival.
The five-year overall survival was 18% for NF1
associated and 40% for sporadic MPNST (de
Vasconcelos, et al., 2017). Our patient died six
months after diagnosis in spite of a surgical
resection and chemotherapy. She had NF1 status and
huge tumor size (13 cm x 9 cm x 9 cm) that
associated with poor prognostic.
Malignant Peripheral Nerve Sheath Tumor in Neurofibromatosis Type 1 Patient
361

4 CONCLUSION
Patients with NF1 need careful follow up because of
the possibility of hidden carcinomas such as MPNST
underneath the neurofibromas. Clinicians should be
alert to unexpected growth of a pre-existing
neurofibroma, particulary a plexiform neurofibroma,
or the occurrence of unexplained pain with/without
neurological symptoms such as weakness and
paresthesias. Fluorodeoxyglucose PET-CT should be
used in suspicious lesion with the potential for
malignant transformation in NF1. Early diagnosis of
MPNST is mandatory to increasing the successful of
surgical resection
REFERENCES
Batista, P. B., Bertollo, E. M. G., Costa, D. de S., Eliam,
L., Cunha, K. S. G., Cunha-Melo, J. R., Rodrigues, L.
O. C., 2015. Neurofibromatosis: part 2 – clinical
management. Arquivos de Neuro-Psiquiatria, 73(6),
531–543. https://doi.org/10.1590/0004-
282X20150042.
Carroll, S.L., 2016. The Challenge of Cancer Genomics in
Rare Nervous System Neoplasms. The American
Journal of Pathology 186, 464–477.
doi:10.1016/j.ajpath.2015.10.023.
Cunha, K.S.G., Caruso, A.C., de Faria, P.A.S., da Silva,
L.E., Pires, A.R.C., Geller, M., et al., 2012. Malignant
peripheral nerve sheath tumors: clinic-pathological
aspects, expression of p53 and survival. Clinics, 67(8),
963-968.
Dunn, G.P., Spiliopoulos, K., Plotkin, S.R., Hornicek, F.J.,
Harmon, D.C., Delaney, T.F., Williams, Z., 2013.
Role of resection of malignant peripheral nerve sheath
tumors in patients with neurofibromatosis type 1.
Journal of neurosurgery 118, 142–148.
doi:10.3171/2012.9.JNS101610.
de Vasconcelos, R.A.T., Coscarelli, P.G., Alvarenga, R.P.,
Acioly, M.A., 2017. Malignant peripheral nerve sheath
tumors with and without neurofibromatosis type 1.
Arq Neuropsiquiatr, 75(6), pp. 366-71.
Farid, M., Demicco, E.G., Garcia, R., Ahn, L., Merola,
P.R., Cioffi, A., Maki, R.G., 2014. Malignant
peripheral nerve sheath tumors. The oncologist 19,
193–201.doi:10.1634/theoncologist.2013-0328.
Friedrich, R.E., Hartmann, M., Mautner, V.F., 2007.
Malignant Peripheral Nerve Sheath Tumors (MPNST)
in NF1-affected children, in: Anticancer Research. pp.
1957–1960.
Goldblum. J.R., Folpe, A.L., Weiss, S.W., 2014. Enzinger
& Weiss’s soft tissue tumors. 6
th
Ed. Philadelphia:
Elseiver Saunders. p. 855-79.
Hwang, I.K., Hahn, S.M., Kim, Hyo Sun, Kim, S.K., Kim,
Hyo Song, Shin, K.-H., Suh, C.O., Lyu, C.J., Han,
J.W., 2016. Outcomes of Treatment for Malignant
Peripheral Nerve Sheath Tumors: Different Clinical
Features Associated with Neurofibromatosis Type 1.
Cancer research and treatment : official journal of
Korean Cancer Association 1–10.
doi:10.4143/crt.2016.271.
Kim, A., Stewart, D.R., Reilly, K.M., Viskochil, D.,
Miettinen, M.M., Widemann, B.C., 2017. Malignant
Peripheral Nerve Sheath Tumors State of the Science:
Leveraging Clinical and Biological Insights into
Effective Therapies. Sarcoma 2017, 7429697.
doi:10.1155/2017/7429697.
Kolberg, M., Høland, M., Agesen, T.H., Brekke, H.R.,
Liestøl, K., Hall, K.S., Mertens, F., Picci, P., Smeland,
S., Lothe, R.A., 2013. Survival meta-analyses for
>1800 malignant peripheral nerve sheath tumor
patients with and without neurofibromatosis type 1.
Neuro-Oncology 15. doi:10.1093/neuonc/nos287.
Korf, B.R., 2000. Malignancy in Neurofibromatosis Type
1. The Oncologist 5, pp. 477–485.
doi:10.1634/theoncologist.5-6-477.
Leroy, K., Dumas, V., Martin-Garcia, N., Falzone, M.-C.,
Voisin, M.-C., Wechsler, J., Revuz, J., Créange, A.,
Levy, E., Lantieri, L., Zeller, J., Wolkenstein, P.,
2001. Malignant peripheral nerve sheath tumors
associated with neurofibromatosis type 1: A
clinicopathologic and molecular study of 17 patients.
Archives of Dermatology 137, pp. 908–913.
Pourtsidis, A., Doganis, D., Baka, M., Bouhoutsou, D.,
Varvoutsi, M., Synodinou, M., Giamarelou, P.,
Kosmidis, H., 2014. Malignant peripheral nerve sheath
tumors in children with neurofibromatosis type 1.
Case Reports in Oncological Medicine 2014, 1–7.
doi:http://dx.doi.org/10.1155/2014/843749.
Zehou, O., Fabre, E., Zelek, L., Sbidian, E., Ortonne, N.,
Banu, E., Wolkenstein, P., Valeyrie-Allanore, L.,
2013. Chemotherapy for the treatment of malignant
peripheral nerve sheath tumors in neurofibromatosis 1:
A 10-year institutional review. Orphanet Journal of
Rare Diseases 8. doi:10.1186/1750-1172-8-127.
RCD 2018 - The 23rd Regional Conference of Dermatology 2018
362
