
Combinatorial Identification of Broad Association Regions
with ChIP-seq Data
Jieun Jeong
1
, Mudit Gupta
2
, Andrey Poleshko
2
and Jonathan A. Epstein
2
1
Gastrointestinal Unit and Center for Computational and Integrative Biology, Massachusetts General Hospital,
Harvard Medical School, Boston, MA 02114, U.S.A.
2
Department of Cell and Developmental Biology, Institute for Regenerative Medicine, and the Penn Cardiovascular
Institute, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, U.S.A.
Keywords: ChIP-seq Data, Regions of Protein Binding, Global Optimization.
Abstract: Motivation: Differentiation of cells into different cell types involves many types of chromatin modifications,
and mapping these modifications is a key computational task as researchers uncover different aspects of that
process. Modifications associated with heterochromatin formation pose new challenges in this context
because we must define very broad regions that have only a moderately stronger signal than the rest of the
chromatin. Lamin-associated domains (LADs) are a prime example of such regions. Results: We present
Combinatorial Identification of Broad Association Regions (CIBAR), a new method to identify these types
of broad regions. CIBAR is based on an efficient solution to a natural combinatorial problem, which adapts
to widely variable yields of reads from ChIP-seq data and the associated controls and performs competitively
with previous methods, including DamID, which has been used in many publications on LADs but cannot be
applied in most in vivo situations.
1 INTRODUCTION
It is now widely accepted that there are many types of
epigenetic chromatin modifications that profoundly
impact cellular phenotypes and thus all types of life
processes (Berstein et al, 2010). A typical task in
processing genome-wide data regarding such
modifications is to simplify the complex distribution
of these modifications into simple collections of
regions where they are present.
There is no universal gold standard concerning
present/absent decisions, but in general, we wish to
see reproducible results (thus removing or decreasing
the impact of data noise) that are associated with
biological outcomes of interest, such as activating or
repressing gene expression or the presence of other
modifications.
Because different modifications are associated with
diverse molecular mechanisms, they require different
computational tools. The best-investigated “regions
of presence” are so-called (narrow) peaks of
sequence-specific transcription factors, which can
have lengths around 100 bps. Broad peaks, with
lengths ranging from hundreds to thousands of bps,
are typical regions for the presence of chromatin
factors, e.g., chromatin-modifying enzymes that form
protein complexes with transcription factors but have
some mobility that allows them to modify longer
chromosomal intervals. A prime example of regions
with peaks and broad peaks are so-called promoters
and enhancers (Ernst et al., 2011), and even longer
broad peaks may be associated with regions with
Polycomb repression complex activity (Ernst et al.,
2011; Mikkelsen et al., 2007). In this note, we focus
on broad regions, which sometimes cover many
millions of base pairs, that in many cases are
associated with modifications that repress gene
expression in wide domains because they are
involved in the establishment of heterochromatin.
The tools for finding broad peaks are not adequate
for finding broad regions because they share the basic
assumption with tools for narrow peaks: namely, they
define locations that have a much higher normalized
concentration of ChIP reads than so-called
background locations. However, heterochromatin
covers most of the genome, and the modifications
associated with heterochromatin have a very wide
distribution; therefore, assumptions of that kind are
not applicable.
One type of chromatin modification that is attracting
Jeong, J., Gupta, M., Poleshko, A. and Epstein, J.
Combinatorial Identification of Broad Association Regions with ChIP-seq Data.
DOI: 10.5220/0005663400270034
In Proceedings of the 9th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2016) - Volume 3: BIOINFORMATICS, pages 27-34
ISBN: 978-989-758-170-0
Copyright
c
2016 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
27
increasing attention is binding to lamins proteins
present in the nuclear lamina, as well as the lamina of
the nucleolus (Padeken and Heun, 2014).
The lamina lines the membranes of the nucleus and
nucleolus and forms a mesh-like structure that
stabilizes the position of a portion of the chromatin in
the nuclear periphery. Chromosomal regions where
contact with lamins is detected are called lamin-
associated domains (LADs); binding to lamins is
mediated through several proteins, and it is still under
investigation. The following proportions have been
reported in the mouse genome: (1) ca. 65% of
chromatin is heterochromatin (Ernst et al., 2011), (2)
ca. 40% of chromatin is in LADs (Guelen et al.,
2008), and (3) more than 90% of LADs are in
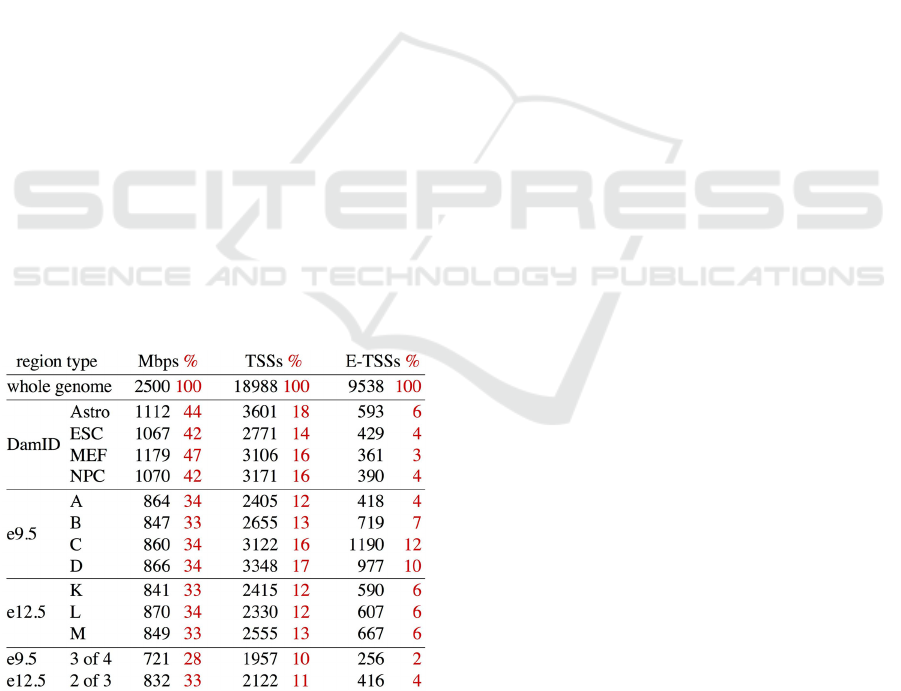
heterochromatin (this estimate is based on the
depletion of LADs with expressed genes [Table 1]).
It is important to note that eukaryotic genomes vary
widely in length and thus in gene density; hence,
those proportions should be different in different
species. The extent of heterochromatin changes
during cell differentiation, and these changes are part
of the processes that determine cell fate. In turn, a
number of chromatin factors have been implicated in
the formation of LADs.
Table 1: Joint length, the number of gene transcription start
sites (TSS) and the number of TSSs of expressed genes (E-
TSS, genes with an e9.5 expression level above the median)
for LADs identified by DamID and by CIBAR. We define
the consensus of the four e9.5 samples (three e12.5
samples) as windows that are selected to be in LADs for all
of them, with one possible exception, and we call the
resulting set "3 of 4" ("2 of 3" for e12.5 samples).
Computation of LADs includes the following three
basic stages, and for each stage we can choose a
method: (1) we obtain the data from the genome
(signal and control), (2) we convert the data to a
"normalized signal", and (3) we use the normalized
signal to determine regions where the normalized
signal values are mostly higher than in the remainder
of the genome.
There are two methods of collecting the binding
data for lamins and proteins that interact with lamins
for the purpose of genome-wide mapping of LADs.
The first is DamID (Guelen et al., 2008). In DamID,
the protein binding data are obtained by adding a
DNA methylating domain to the investigated protein.
Then, the loci with methylated DNA are identified by
hybridization to microarrays (with ca. 2.5 million
probes in the case of the mouse genome). These data
are paired with data in which the protein is augmented
with a “neutral” domain. The benefits are that no
specific protein antibodies are required and that the
control data match the signal data very well.
However, such genomic manipulation is impractical
in vivo; in particular, DNA methylation itself can
affect the phenotype. The second method is to collect
ChIP-seq using an antibody and input, which is the
associated control formed from a portion of the
starting material, i.e., the DNA from the respective
cell sample. The signal data that are collected are
quite uniformly distributed while methods of the
broad peak-type perform well only when we have a
high fold change between regions where we declare
the present compared with the remainder of the
chromatin.
The standard method for computing the
“normalized signal” from the data is to use ratios,
such as the “the number of ChIP reads” over “the
number of input reads.” However, we present our
alternative method in Section 2.
Several methods have been previously used to
identify broad regions from binding data.
The first method is a 2-state Hidden Markov
Model (HMM). The use of HMM for defining
genomic regions is well established (e.g., Ernst et al.,
2011) and seems indispensable when we integrate the
signals of multiple types. HMM has been used by Xu
et al. (2010) to compute the broad peaks from ChIP-
seq/input data, and is also a part of the DamID
method. However, when we have only one type of
signal, combinatorial algorithms are faster and offer a
better understanding of how the regions are defined,
and consequently, of how to set the parameters to
achieve the desired outcomes.
Lund et al. (2014) have tested several existing
programs for computing “long peaks” and have found
that all of them are inadequate for LADs. Another
combinatorial approach used to identify LADs is the
“sliding windows” method (Shah et al., 2013), in
which a long window consists of k short windows
(called steps). We accept a long window if it contains
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
28
some threshold number of short windows with the
positive score, and we declare LADs as the union of
the accepted long windows. An undesirable aspect of
this method is that it ignores the signal values that can
have considerable amplitudes and are not captured by
comparison with a threshold. Moreover, the user must
arbitrarily choose many parameters, including the
threshold on the values, the length of the long
windows and the minimum number of “positive”
short windows.
Those undesirable aspects are not present in a
method that uses a combinatorial problem defined in
terms of the sums of signal values (that can be
positive and negative) in the selected intervals. The
EDD method by Lund et al. (2014) incorporates the
following combinatorial problem: (1) we are given a
number of arrays with real values and (2) we find a
contiguous fragment with the maximum sum of
values. Then, we select that fragment and remove it
from its array, which may increase the number of
remaining arrays. We repeat this selection a desired
number of times, and finally we reject the selected
regional candidates that do not pass a permutation test
for significance.
Our CIBAR method has two major differences
from EDD. First, we use a different formula for
computing the signal values that more accurately
follows the dependency between the distribution of
the ChIP and input reads. Second, instead of selecting
the fragments one at a time, we apply a framework of
global optimization such that for a given array and k,
we select k array fragments in such a way that the sum
of all of the values in the selected fragments is
maximized. We also show a very efficient solution to
that problem.
2 METHODS - DESCRIPTION
OF CIBAR
2.1 Windows
We partitioned the genome into 1000 bp windows
(also known as bins) and each window has a position,
e.g., chr2:3001 for bps 3,001,001 to 3,002,000. Each
window w has a
w
input reads and b
w
ChIP reads.
Because the windows are merged into much longer
regions, the precise selection of the window length
has moderate importance. However, in our
experience, the average value of the a
w
, number of
input reads in a window should be in the range of 50.
We collected reads from several ChIP and input
sets, and we process only windows with at least one
read mapped in one of the sets. The other windows
are ignored, and we call them “null windows.” As a
result, we do consider windows with a
w
= b
w
= 0 if
they have reads in some other data sets.
2.2 Scores of Windows – The
Motivation
The “significance” of an interval of windows is
related to the p-value that we see for a particular
number of signal reads in that interval, but we
normalize that number on the basis of the number of
input (control) reads.
The question that we address in a novel way is
how to normalize the signal. ChIP reads are not
uniformly collected through the genome because of
the differences in the accessibility of different parts
of chromatin to the processes that generate the reads
(Kharchenko et al. 2008). In particular,
heterochromatin tends to be less accessible, and the
frequency of input reads in heterochromatin tends to
be lower. This motivates normalization approaches,
such as the ratio between the normalized ChIP and
input read counts.
The ratio approach is valid if the relationship
between the ChIP and input counts is linear.
However, this is not the case.
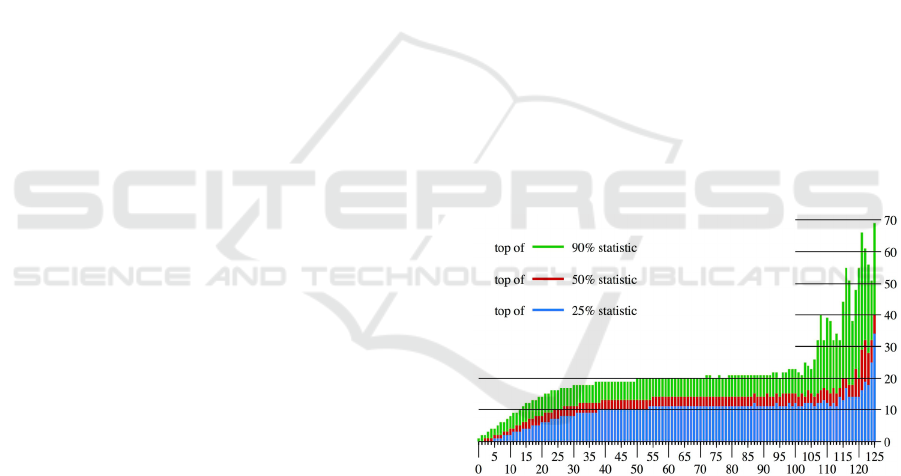
Figure 1: The X-axis is the a
w
, which is the number of input
reads in a 1 kbp window, and each bar represents the set of
all windows with a
w
indicated by the X-coordinate. The
heights of the colors for the Y-coordinate are the values of
b
w
, which are the numbers of ChIP reads that are order
statistics for this window set. The top of blue is 25% (the
highest value in the bottom quadrant), the top of red is 50%
(the median), and the top of green is 90% (the lowest value
in the top decile). Forty-four is the median level for the
input reads, less than 0.1% of the windows have more than
100 input reads, and because the sets for a
w
> 100 are very
small, their statistics change quite irregularly.
Figure 1 illustrates the distribution for a typical
input-ChIP pair. In that example, the input-ChIP
relationship is well approximated as linear between 0
Combinatorial Identification of Broad Association Regions with ChIP-seq Data
29

and 20 input reads, and as constant between 30 and
100 input reads; i.e., the ChIP read counts are almost
independent of the input counts. For rare high counts
of input reads, the relationship is once again roughly
linear.
Normalization is important because 20-25% of
windows have low input counts and proportionally
smaller ChIP counts. Moreover, these windows are
primarily in the heterochromatin where we expect
LADs to be. However, for a wide range of input
values, linear normalization would be a significant
distortion. Our solution is to normalize the signals
separately for each possible number of input reads.
To our knowledge, previous work has made no
account of this problem. The use of ChIP-input ratio
did not produce very distorted results because the
effect is to exaggerate the link between LADs and
heterochromatin, although in actuality, this link is
very strong. However, in studies of embryonic
differentiation, etc., we are very much interested in
“marginal” LADs, where the dynamic extent of
LADs has regulatory impact on gene expression
without total heterochromatin silencing
characteristics. Thus, misclassification of LADs, over
1-5% of the genome can have a high impact on the
lists of genes whose regulation can be attributed to
LADs. Moreover, the positive bias for
heterochromatin can be detrimental when the method
is applied to regions associated with PRC activity or
gene elongation.
Other than separating the windows according to
their input counts, our method for computing the
signal is quite similar to the one by Lund et al. (2014).
2.3 Window Scores – The Formula
Given that a window w has a
w
input reads and b
w
ChIP
reads, we define the following statistics:
(1) n
i
is the number of all of the windows with a
w
= i,
(2) s
ik
is the number of all of the windows with a
w
= i and b
w
= k, and
(3) A
ik
is the average rank of windows with a
w
= i
and b
w
= k among all of the windows with a
w
= i, i.e., A
ik
= s
i0
+ … + s
i(k-1)
+ ½s
ik.
The score(i, k) = κ(2
A
ik
/n
i
), where
κ(x) = log x if x ≤ 1
and κ(x) = −log (2−x) if x ≥ 1.
Note that the score(i, k) is negative/zero/positive if A
ik
is smaller/equal/greater than ½n
i
, respectively.
The idea of the scoring is that we prefer to select
windows with positive scores and to avoid selecting
windows with negative scores. However, with the
described method for assigning scores, about half of
the windows have positive scores, and about half have
negative scores, even though the target regions
occupy less than half of the genome. Therefore, we
use
score(w) = score(a
w
, b
w
) – α,
where α is the score adjustment, which is the same for
all of the non-null windows. The larger the score
adjustment we use, the fewer the windows belonging
to the selected regions. The score adjustment is one
of the two parameters supplied by the user.
This scoring formula is subjected to a high
statistical fluctuation when n
i
is small. If n
i
is smaller
than 1000, we enlarge the counts by forming a set of
windows of size 1000 or more, which have between
i−d and i+d input reads for the smallest possible d.
2.4 From Signal Values to Regions/
Domains
LAD signals computed with our formula exhibit large
random-like variability, in part because the collection
of reads, both input and ChIP, is essentially a random
process of extracting the reads from a library. In the
case of narrow and broad peaks, we observe
concentrations of ChIP reads that are many times
larger than background, although this is not the case
with the much less-concentrated signal that we see in
very broad regions. Instead, we can see a rather small
advantage of the positive signal that persists over a
long interval.
We identify our broad regions by solving the 1-
dimensional fragment selection (1DFS): given an
array with real values (scores of windows in
chromosomes) and a target number k, find k
disjointed fragments that are contiguous sub-arrays
and have the maximum sum of scores. The 1DFS
problem describes the optimization goals more
directly than the iterative formulation by Lund et al.
(2014). Importantly, exact solution of 1DFS can be
found efficiently in time proportional to n log n,
where n is the number of windows. This is slightly
slower than the speed of the algorithm in Lund et al.
(2014), but the probabilistic validation of the
computed regions takes approximately 10 times
longer and our entire computation takes about one
minute on a 1.7 Ghz Macintosh computer. Our
algorithm for 1DFS uses a greedy method and is easy
to implement.
We give the details of the 1DFS algorithm and the
proof of its correctness in the Appendix. To our
knowledge, this is a new algorithm that may have
more applications in genomic region studies.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
30
3 RESULTS
We applied our method to laminB1 ChIP data
collected for the following two biological conditions
(ChIP was performed as described in Shah et al.
(2013)): (1) the mouse embryonic heart at e9.5 and
(2) the mouse embryonic heart at e12.5 (days after
conception), with four and three replicates,
respectively. These samples are subsequently referred
to as A-D and K-M, respectively. We found that using
an input with less than 50 million reads leads to erratic
results, so we used the same input for all of the ChIP
samples; it was collected from the e9.5 sample and
had 111 million reads. We set the target number of
LADs at 1500 and experimented with different
adjustment parameters. Adjustments that led to LADs
having much more than 800 Mbps in joint length had
a considerable proportion of regions with p-values
above 0.001, so we decided not to further increase the
size (see Appendix for the details of p-value test).
Then, we checked whether the LADs have the
properties previously reported in the literature. The
first property is that LADs are severely depleted of
genes and that the genes present in LADs are mostly
silent (very low expression level). We tested this on
18,988 genes that have annotations in refGene.txt for
the mm9 mouse genome build (available from UCSC
Genome Browser) and for which we also had
microarray expression data.
Table 1 shows that our LADs contain similar
numbers of genes and expressed genes as LADs
computed with the DamID method (see Peric-Hupkes
et al., 2010). DamID LADs cover 42-47% of the
genome, 14-18% of genes and 4-6% of expressed
genes. Our LADs (identified by CIBAR) cover 33-
34% of the genome, representing proportionally
fewer genes, and are also depleted expressed genes,
though not as much.
Taking a consensus of regions computed for the
same condition results in a similar depletion in
expressed genes as in DamID; consensus is an
effective method to eliminate false positives. We
defined the consensus as the base pairs that occur in
all or all but one of the LAD sets that were computed
for that condition; this is shown in Tables 1-3.
We also measured the level of consistency for the
LADs computed for the four cell types studied in
Peric-Hupkes et al. (2010) and our LADs from the
embryonic heart (see Table 2).
The degree of consistency between our embryonic
LADs and the LADs computed in Peric-Hupkes et al.
(2010) is similar to the consistency between those cell
types (note that among those four types, neural
pluripotent cells (NPC) and astrocytes, a
differentiated neural type, are the closest).
Table 2: Similarity of different LAD sets. For e9.5 and
e12.5, we use consensus regions (called “3 of 4” and “2 of
3”). The percentage of the smaller set that belongs to both
sets being compared is in red.
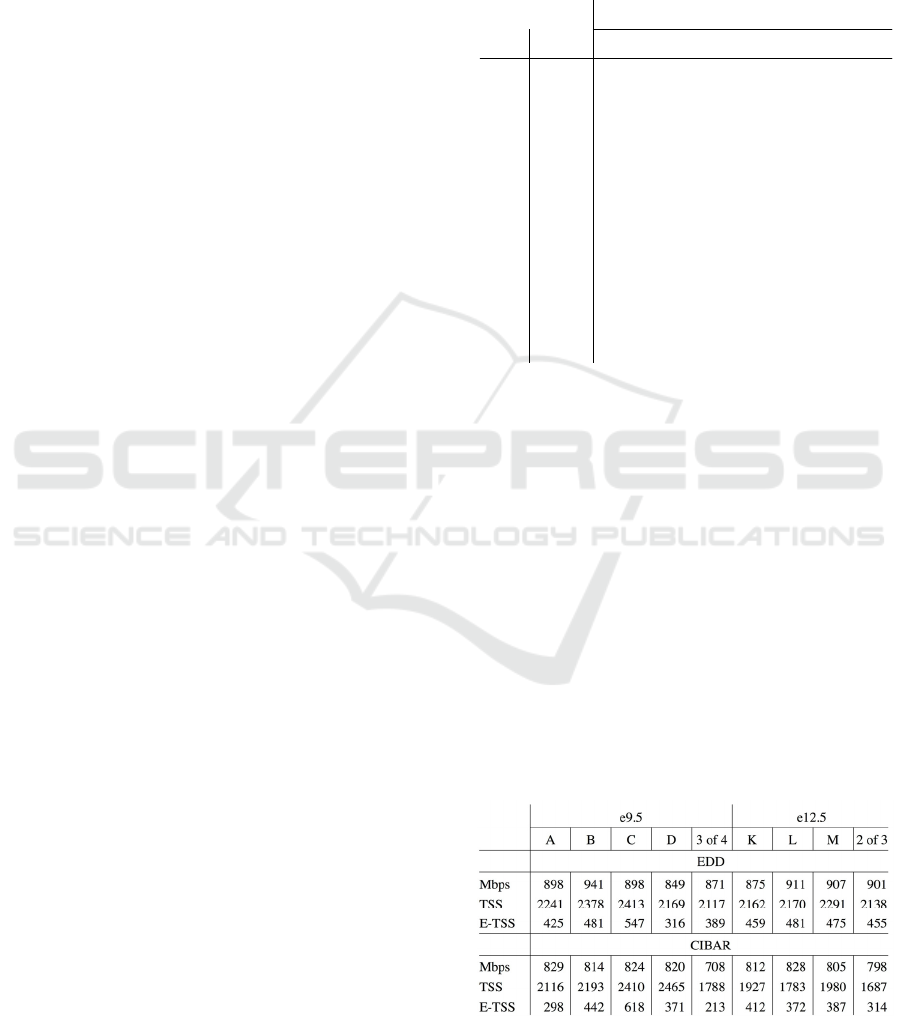
In Table 3 we compare EDD with CIBAR.
Both EDD and CIBAR produce LADs with
properties reported in Peric-Hupkes et al. (2010),
namely that they cover approximately 40% of the
genome (32-33% in the case of CIBAR and 34-36%
in the case of EDD) and that LADs are depleted in
genes, even more so in expressed genes, as we already
discussed. The consistency with DamID LADs is also
similar. CIBAR LADs have two advantages. First,
they are computed with 1 kbp windows, while EDD
automatically selects window sizes between 35 and
90 kbps, which means that the LAD boundaries are
much less precise. Second, EDD produced 200-275
LADs, and CIBAR produced 880-1280 LADs, which
is closer to what was previously reported.
Table 3: Comparison of results of CIBAR and EDD. The
column labels correspond to row labels in Table 1.
To a degree, this is a consequence of the parameters
we chose for CIBAR, namely the selection of the
Common Mbps % of the smaller set
Set Mbps e9.5 e12.5 Astro ESC MEF NPC
e9.5 721 565 584 581 600 594
78 81 81 83 82
e12.5 832 565 577 605 635 579
81 69 73 76 70
Astro 1112 582 577 772 881 931
81 69 72 78 87
ESC 1067 581 605 772 836 786
81 73 72 78 87
MEF 1179 600 635 881 836 880
83 76 78 78 82
NPC 1070 594 579 931 786 880
82 70 87 74 82
Combinatorial Identification of Broad Association Regions with ChIP-seq Data
31

window size, target size (regulated by the adjustment)
and the target number of regions.
4 CONCLUSIONS
We presented a new method of computing broad
regions associated with chromatin modifications that
is applicable even when ChIP data do not exhibit
large fold changes between the affected regions and
the rest of the genome. Although our method was
conceived and implemented before the publication of
EDD (Lund et al., 2014), it shares the following three
aspects: (a) using scores of windows rather than a
selected cut-off between “good” and “bad” windows,
(b) basing the score number on the ranks of the
windows and (c) applying a natural combinatorial
problem to group windows into regions.
Our scoring method models the distributions of
ChIP and control reads more accurately; thus, we
avoid the positive bias for selecting windows in the
least accessible parts of the genome. It remains an
open question whether this is a good model, and we
expect further progress in this direction.
The combinatorial problem that we have
applied, 1DFS, is much more natural than the iterative
selection of fragments with the highest sum of scores,
which can excessively merge “positive” regions with
the “negative” regions that separate them. Lund et al.
(2014) introduced gap penalty (decreasing all
negative scores by a constant) to reduce that
tendency, but we suspect that this is one of the reasons
why EDD works with such low granularity. Although
1DFS is a global optimization problem, we have
found a solution that is very efficient and easy to
implement.
Our method uses only two parameters, k and α,
but the proper selection of parameters remains an
open problem. In Section 2, we set k to obtain the
number of LADs, which is close to the number
reported in papers applying the DamID method (see
Peric-Hupkes et al., 2010). Parameter α can be
selected in different ways. As it increases, the
proportion of windows with positive score(w)
decreases, as does the sum of lengths of identified
LADs. However, when we decrease α too much, the
p-values of the computed LADs tend to increase, and
we cannot suggest a statistic that allows to optimize
α. In fact, we tested our program and EDD on six
genes confirmed to be in LADs using ChIP-qPCR
(data not included). We found that increasing α may
paradoxically exclude some of them, whereas
choosing a consistent α of 0.12 led to consistent
inclusion of 5 of the genes in LADs computed for all
four e9.5 samples. LADs computed by EDD (which
automatically adjusts parameters to optimize the p-
values) consistently included exactly 3 of these genes.
This small evidence suggests that at present there is
no better way to select the parameters than using
whatever knowledge we have, most preferably some
genomic positions confirmed to be in LADs or
outside LADs, and picking the parameters to be
consistent with that knowledge. The situation with
identifying short peaks of transcription factors is
similar because the existing programs can produce
"false positives," i.e., statistically significant peaks
that are too weak to have a biological impact.
Therefore, these programs provide options to select
the parameters, such as maximum p-value/FDR,
minimum fold change.
REFERENCES
Bernstein BE et al. (2010) The NIH Roadmap Epigenomics
Mapping Consortium. Nature Biotechnol. 28(10),
1045-8.
Ernst, J. et al. (2011) Mapping and analysis of chromatin
state dynamics in nine human cell types. Nature,
473(7345), 43–49.
Guelen, L. et al. (2008) Domain organization of human
chromosomes revealed by mapping of nuclear lamina
interactions. Nature, 453(7197), 948–951.
Kharchenko, P. V. et al. (2008) Design and analysis of
ChIP-seq experiments for DNA-binding proteins. Nat.
Biotechnol., 26(12), 1351–1359.
Lund, E. et al. (2014) Enriched domain detector: a program
for detection of wide genomic enrichment domains
robust against local variations. Nucleic Acids Res.,
42(11), e92.
Mikkelsen, T. S. et al. (2007) Genome-wide maps of
chromatin state in pluripotent and lineage-committed
cells. Nature, 448(7153), 553–560.
Padeken, J. and Heun, P. (2014) Nucleolus and nuclear
periphery: velcro for heterochromatin. Curr. Opin.
Cell. Biol., 28, 54–60.
Peric-Hupkes, D. et al. (2010) Molecular maps of the
reorganization of genome-nuclear lamina interactions
during differentiation. Mol. Cell., 38(4), 603–613.
Shah, P. P. et al. (2013) Lamin B1 depletion in senescent
cells triggers large-scale changes in gene expression
and the chromatin landscape. Genes. Dev., 27(16),
1787–1799.
Xu, H. et al. (2010) A single-noise model for significance
analysis of ChIP-seq with negative control.
Bioiformatics, 26(9), 1199–1204.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
32

APPENDIX – THE GREEDY
ALGORITHM FOR THE 1DFS
PROBLEM
1.1 Problem Definition
An instance of 1DFS consists of an array of real
values A[0] … A[n−1] and a positive integer k. A
fragment A[s, t] is specified by two distinct integers;
if s < t then it consists of A[s] … A[t−1], otherwise it
is a “wrap around” fragment that consists of A[s] …
A[n−1] and A[0] … A[t−1]. The value of a fragment
is the sum of its entries. The goal is to find j ≤ k
disjointed fragments with the maximum possible sum
of values.
Allowing the wrap-around fragments simplifies
the problem such that every entry has two neighbors,
left and right, that can join it in the same fragment.
Moreover, it is equivalent to find j disjoint and non-
adjacent fragments with the maximum sum of values
and to find j fragments with the minimum sum of
values because the complement of j disjoint and non-
adjacent fragments also consists of j disjoint and non-
adjacent fragments.
In our application, we have a number of arrays,
one for each chromosome. However, even though
1DFS has only one array and allows wrap-around
fragments, it is in fact equivalent. We can add an entry
with a very low negative value to the end of each of
those arrays and combine all of them into one. Then,
the “improper” fragments are formally allowed, but
they cannot be present in an optimal solution because
they have negative values.
1.2 Instances with Restricting
Partitions and the Greedy
Algorithm
We generalize problem instance by adding P, a
restricting partition of array A, into disjoint
fragments, and then we allow only fragments that are
unions of fragments from P in the solution. The
fragments from P can be numbered p
0
… p
m
−1
, and
we use A
P
[i] to denote the value of p
i
.
Our algorithm starts with the restricting partition of
A into 1-entry fragments and then applies the rules
described in Figure 2 until it terminates. The
algorithm is greedy in the sense that the rules are very
simple, and it either terminates or applies a rule that
reduces the size of the problem (the number of
fragments in the restricting partition).
Use the first applicable rule from the following list:
(1) Terminating Rule. If P contains k or fewer
fragments with positive values, return the set of
these fragments.
(2) Equal Sign Rule. If p
i
and p
i+1
both have
negative values or both have non-negative values,
merge them together.
(3) Minimum Value Rule. Select i with the
minimum | A
P
[i] |, merge together p
i-1
, p
i
and p
i+1.
Figure 2. The rules in the algorithm for the 1DFS
problem.
1.2.1 Correctness of the Terminating Rule
The sum of the values from the selected fragments
cannot exceed the sum of the values from all of the
positive fragments in P; thus, the value of the returned
solution is optimal, and because there are at most k
such fragments, the solution is valid.
1.2.2 Correctness of the Equal Sign Rule
Suppose that the solution S is consistent with the
former restricting partition but not with the new one.
Then, a fragment of S (F, for example) contains p
i
but
excludes p
i
+1
or vice versa. If the value of p
i
is
negative, we can modify F by excluding p
i
, thus
increasing the value of F and S; this works because p
i
is at the end of F. Similarly, if the value of p
i
is non-
negative, we can modify F by extending it with p
i
+1
,
which before was fully excluded from the solution,
and the value of the solution will increase or stay the
same because the value of p
i
+1
is also non-negative.
Thus we defined a solution S’ that has the same value
as S and is consistent with the new partition.
1.2.3 Correctness of the Minimum
Value Rule
Because the Termination Rule does not apply, the
number of the positive restriction fragments is l > k.
Because the Equal Sign Rule does not apply, the
fragments of P with negative and non-negative values
alternate, so the number of P-fragments is equal to 2l
> 2k.
Suppose that solution S is consistent with the
former restricting partition, P, but not with the new
one P’. This means that the fragments of S contain
some but not all of p
i
−1
, p
i
and p
i
+1
. We can assume
that S cannot be improved by extending or shrinking
one of its fragments by one P-fragment. Hence, in
every fragment of S, the first and the last P-fragment
have positive values, and in every fragment of the
complement of S, the first and last P-fragment has
negative values. Thus, each fragment of S and of the
Combinatorial Identification of Broad Association Regions with ChIP-seq Data
33

complement of S consists of an odd number of
restriction fragments.
Case 1: Fragments of S contain only p
i
−1
. We can
extend S by incorporating p
i
and p
i
+1
, and the value of
S increases by A
P
[i+1]−|A
P
[i]| ≥ 0.
Case 2: Fragments of S contain only p
i
, so the
restriction fragment p
i
is also a fragment of S. We
remove p
i
from S, which decreases the value of S by
x, which is the value of p
i
. We have to modify S to
increase its value by x or more. Because we have
decreased the number of fragments in S, the
modification is allowed to increase the number of
fragments by 1. Because there are more than 2k
restriction fragments, one of the fragments of S or the
complement of S (F, for example) contains at least
three restriction fragments. If F is a fragment of S, it
contains a restriction fragment p
j
with a negative
value –y. We remove p
j
from F, which increases the
number of fragments by one and increases the value
of the solution by y ≥ x. If F is a fragment of the
complement of S, it contains a restriction fragment p
j
with a positive value y, so we add p
j
to S. Again, the
number of fragments in S increases by 1 and the value
by y≥x. Importantly, as p
j
neither starts nor ends F,
it cannot be p
i
−1
or p
i
+1
.
Other cases are symmetric, so every solution that
satisfies the restriction before we applied the merging
can be modified to a solution that satisfies the new
restriction with a value that is the same or larger.
Implementation
We first compute a matrix with read counts for every
data sample (input and ChIP) and every window, and
we then use that matrix (tab separated file) as the
input for our C-program. Null windows, those
without any reads, are ignored as “unmappable”.
In the program, we use an array of doubly linked
lists for fragments in the restriction partition, with one
list for each chromosome. The fragments are also
represented in a binary heap (priority queue), which
allows rapid application of the Minimum Value Rule.
To assess the p-value of computed regions, we
used the null model in which the scores of windows
are randomly permuted. We found out that for regions
with more than 200 windows, the p-value can be well
approximated using the cumulative Gaussian
distribution. For fewer windows, we use one million
runs of 200 random window selections, which
allowed us to tabulate empirical p-values for all of the
region sizes up to 200 windows. As a result, this
Monte Carlo test adds less than a minute to the
computation time and uses very little additional
memory.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
34
