
Alpha Complexes in Protein Structure Prediction
Pawel Winter and Rasmus Fonseca
Department of Computer Science, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen Ø, Denmark
Keywords:
Protein Structure Prediction, Force Field, Alpha-complexes, Kinetic Data Structures.
Abstract:
Reducing the computational effort and increasing the accuracy of potential energy functions is of utmost
importance in modeling biological systems, for instance in protein structure prediction, docking or design.
Evaluating interactions between nonbonded atoms is the bottleneck of such computations. It is shown that
local properties of α-complexes (subcomplexes of Delaunay tessellations) make it possible to identify non-
bonded pairs of atoms whose contributions to the potential energy are not marginal and cannot be disregarded.
Computational experiments indicate that using the local properties of α-complexes, the relative error (when
compared to the potential energy contributions of all nonbonded pairs of atom) is well within 2%. Further-
more, the computational effort (assuming that α-complexes are given) is comparable to even the simplest and
therefore also fastest cutoff approaches.
The determination of α-complexes from scratch for every configuration encountered during the search for the
native structure would make this approach hopelessly slow. However, it is argued that kinetic α-complexes can
be used to reduce the computational effort of determining the potential energy when “moving” from one con-
figuration to a neighboring one. As a consequence, relatively expensive (initial) construction of an α-complex
is expected to be compensated by subsequent fast kinetic updates during the search process.
Computational results presented in this paper are limited. However, they suggest that the applicability of α-
complexes and kinetic α-complexes in protein related problems (e.g., protein structure prediction and protein-
ligand docking) deserves furhter investigation.
1 INTRODUCTION
In protein structure prediction a vast atomic configu-
ration space has to be searched when looking for the
native configuration minimizing its potential energy.
Good potential energy estimators require substantial
computational effort. Reducing this effort is therefore
important. Furthermore, similar searches and poten-
tial energy estimations arise in for example protein-
protein docking and in protein design.
Interactions between nonbonded atoms are the
computational bottleneck of potential energy estima-
tions. Commonly used cutoff methods compute the
distances between all pairs of nonbonded atoms and
calculate the contributions of those within some pre-
specified cutoff distance. Different types of contribu-
tions such as van der Waals and Coulomb potentials
may require different cutoff values (Schlick, 2010).
Hierarchical decompositions of proteins with appro-
priately chosen bounding volumes have also been
used to speed up potential energy estimations (Lotan
et al., 2004; Winter and Fonseca, 2012). We show
that α-complexes (which are subcomplexes of well-
known Delaunay tessellations) for appropriately cho-
sen real values of α, α ≥ 0, are well-suited for the
identification of nonbonded pairs of atoms essential
for the estimation of potential energy of proteins. The
identification of such pairs involves exploiting the
structural properties of α-complexes while making
the distance computations for cutoff purposes unnec-
essary. Computational experiments reported in this
paper indicate that the relative error is well within
2% while the computational effort is comparable with
even the simplest cutoff approaches.
Searching for a configuration minimizing the po-
tential energy typically involves perturbing one con-
figuration to obtain the next. It can for example
be achieved by small dihedral rotations of covalent
bonds. As these rotations are carried out, the under-
lying α-complexes can be appropriately updated. We
sketch how these updates can be carried out using the
kinetic data structure framework. In particular, bond
rotations imply that groups of atoms rotate around the
same axis with the same rotational speed on circular
orbits in parallel planes. This significantly speeds up
the computations needed to update α-complexes. Fast
178
Winter P. and Fonseca R..
Alpha Complexes in Protein Structure Prediction.
DOI: 10.5220/0005251401780182
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2015), pages 178-182
ISBN: 978-989-758-070-3
Copyright
c
2015 SCITEPRESS (Science and Technology Publications, Lda.)

updates, in turn, imply fast determination of potential
energy for the next neighboring configuration.
2 SYSTEMS AND METHODS
A force field is a collection of parameters and math-
ematical expressions that together define a function
approximating the potential energy of a system of
atoms. Such a function typically includes bonded
terms capturing forces between covalently bonded
atoms and nonbonded terms capturing forces of non-
bonded atoms.
A simple but still reasonable force field approx-
imating the potential energy (in kcal/mol) of a par-
ticular conformation of a protein (Levitt et al., 1995;
Schlick, 2010) is given by E = E
B
+ E
N
where the E
B
term comprises the contributions from bonded atoms
and the E
N
term comprises the potential energy con-
tributions from nonbonded atoms. It is defined by
E
N
= E
NV
+ E
NC
, where
E
NV
=
∑
p=(i, j)
(e
i j
[
r
e
i j
r
i j
]
12
−2e
i j
[
r
e
i j
r
i j
]
6
)
and
E
NC
= 332
∑
p=(i, j)
q
i
q
j
r
i j
where p is a pair of atoms i and j, r
e
i j
= (r
e
i
+ r
e
j
)/2
with r
e
i
and r
e
j
being van der Waals radii of interacting
atoms, r
i j
is the actual distance between the atoms i
and j, e
i j
=
√
e
i
e
j
with e
i
and e
j
being partial charge
parameters of interacting atoms. Finally, q
i
and q
j
are
partial charges of the atoms i and j.
Nonbonded interactions between atoms separated
by less than three bonds along the covalent structure
are disregarded (Levitt et al., 1995). Their interac-
tions are assumed to be accounted for by bonded in-
teractions.
It is evident that nonbonded terms depend on the
distances between interacting atoms. As the distances
grow, the potential energy contributions become neg-
ligible. In order to speed up the computations, van der
Waals interactions between atoms more than 8-12
˚
A
apart can be disregarded. The cutoff distance for E
NC
is higher (Levitt et al., 1995). Other, more sophisti-
cated cutoff techniques have been suggested (Schlick,
2010).
3 ALGORITHMS
An α-ball b
p
centered at a point p with radius α,
α ≥0, is the set of points at most α away from p. Let
S be a set of n points in the 3-dimensional Euclidean
space E
3
. Let T be a subset of S of size |T | = k + 1,
0 ≤ k ≤3. The convex hull σ
T
of T is also referred to
as a k-simplex. Given a k-simplex σ
T
, any k
0
-simplex
σ
T
0
, T
0
⊂T , is a (proper) face of σ
T
. A k-simplex σ
T
,
0 ≤ k ≤ 3, belongs to the Delaunay complex, (DC )
iff there exists a sphere that passes through the points
of T and all other points of S are strictly outside this
sphere. Assuming the general position (i.e., no five
points lie on a common sphere), the Delaunay com-
plex is a simplicial complex.
Let b
T
denote the smallest ball that passes through
the points of a k-simplex T , 0 ≤ k ≤ 3. Let ρ
T
de-
note the radius of b
T
. If b
T
contains no point of S
in its interior, σ
T
is called Gabriel. Let α be a pos-
itive real number. T is said to be α-short if ρ
T
≤ α.
The α-complex S
α
consists of Delaunay simplices that
are Gabriel and α-short together with their proper
faces (Edelsbrunner, 1992).
Since S
α
1
⊆S
α
2
for α
1
≤α
2
, α-complexes provide
a filtration of a growing family of simplicial com-
plexes, beginning with S
0
(= S) and ending with S
∞
(= DC ). A filtration has O(n
2
) α-complexes since
there are O(n
2
) k-simplices, 0 ≤ k ≤3 (Seidel, 1995).
α-complexes for the protein 2JZC with 224 amino
acids and 3170 atoms and for selected values of α are
shown in Fig. 1.
Figure 1: α-complexes for the protein 2JZC with α =
1.5,2.0,3.0 and ∞.
As α grows, more and more simplices are in-
cluded in S
α
. A simplex σ
T
becomes a member of
S
α
when α = ρ
T
and b
T
is exposed. As α contin-
ues to grow, σ
T
can become a face of another sim-
plex (for example a triangle can become a face of a
tetrahedron). Later on, it can become an interior sim-
plex (for example a face of tetrahedron can become
a face of another tetrahedron). The α-values of these
events can be easily computed from the DC (Edels-
brunner et al., 1998). This is important since in many
applications one is not really interested in S
α
but in
AlphaComplexesinProteinStructurePrediction
179

the simplices on the boundary of S
α
. For a survey
of the applications of α-complexes to various protein-
related problems, see (Zhou and Yan, 2014; Winter
et al., 2009).
We make a simplifying assumption that all atoms
have the same size. Consequently, we focus on α-
complexes of points (or balls with the same radius).
Weighted α-complexes (Edelsbrunner and M
¨
ucke,
1994) and β-complexes (Kim et al., 2006) for spheres
of different sizes have also been developed. However,
these more complicated structures require more com-
putational effort to be constructed and updated.
Consider an α-complex S
α
for some protein and
for a fixed value of α, α > 0. S
α
can also be viewed
as an undirected graph G(α) with the vertices cor-
responding to the atom centers and the edges corre-
sponding to 1-simplices of S
α
. Since the lenghts of
covalent bonds in proteins are assumed to be fixed and
are between 1
˚
A and 1.5
˚
A, G(α) will be connected al-
ready for α ≈ 0.80. For each vertex i, let N
d
(i) de-
note the vertices of G(α) that can be reached from
i by traversing at most d edges. Let N
∗
d
(i) denote the
subset of N
d
(i) containing vertices representing atoms
that are at least three covalent bonds away from i. Let
E
∗
N
= E
∗
NV
+ E
∗
NC
where
E
∗
NV
=
1
2
∑
i∈G(α)
∑
j∈N
∗
d
(i)
(e
i j
[
r
e
i j
r
i j
]
12
−2e
i j
[
r
e
i j
r
i j
]
6
)
and
E
∗
NC
=
332
2
∑
i∈G(α)
∑
j∈N
∗
d
(i)
[
q
i
q
j
r
i j
]
We are interested in estimating relative errors
ε
NV
= |E
∗
NV
−E
NV
|/|E
NV
|
and
ε
NC
= |E
∗
NC
−E
NC
|/|E
NC
|
at different values of α and d.
4 IMPLEMENTATION
The applicability of α-complexes to the determina-
tion of potential energy of proteins was carried out as
follows. In the first stage, 5 proteins (1X5R, 1X0O,
1XDX, 1AKP, 1Y6D) with 110-120 amino acids were
tested. This was done to verify the stability of the ap-
proach as well as to estimate the quality of the solu-
tions obtained. In the second stage, two bigger pro-
teins (2ZJC, 224 amino acids and 3WCZ, 308 amino
acids) were investigated to check if the approach re-
mains robust as the size of proteins increases.
The relative error ε
NV
of the van der Waals contri-
butions to the potential energy of 1X0O remains the
same already for α > 1.6
˚
A, d = 1,2, ...,5. Further-
more, ε
NV
≈ 0%, d = 3,4,5 while ε
NV
≈ 0.22% for
d = 2 and ε
NV
≈ 1.86% for d = 1. Similar relative
errors were observed for the other four proteins with
110-120 amino acids. For the larger proteins 2JZC
and 3WCZ, ε
NV
was also stable for α > 1.6
˚
A. Fur-
thermore, for 2JZC, ε
NV
≈ 0% for d = 3,4, 5 while
ε
NV
≈ 0.19% for d = 2 and ε
NV
≈ 1.74% for d = 1.
For 3WCZ, ε
NV
≈ 0% for d = 3,4, 5 while ε
NV
≈
0.41% for d = 2 and ε
NV
≈ 3.16% for d = 1.
Fig. 2 shows the relative error ε
NC
of the Coulomb
contributions to the potential energy of 1X0O for the
values of α = 1.0, 1.1.,..., 5.9 . It can be seen that
ε
NC
≤2% already for α > 1.6
˚
A and d = 3, 4,5. Also,
ε
NC
≤ 2.5% for d = 2. For d = 1 and α > 1.6,
ε
NC
≈ 6%. Similar relative errors were observed for
the other four smaller proteins with 110-120 amino
acids. For 2JZC with 224 amino acids, ε
NC
≤ 1.5%
for α > 1.6 and for all d = 1,2, ...,5. It is perhaps
somewhat surprising that this was the case for d ≤ 2.
For 3WCZ with 308 amino acids, ε
NC
≤ 1.5% for
α > 1.6 and d = 3,4, 5. For d = 2 and α > 1.6,
ε
NC
≤ 4% while for d = 1 and α > 1.6, ε
NC
≤ 7%.
Fig. 3 shows the time (in ms) needed to compute
van der Waals and Coulomb contributions of non-
bonded atom pairs of 1X0O (using Mac OS X with
1.7 Ghz Intel Core i5 processor). The graph for d = 1
is not shown as it overlaps with the x-axis and is also
covered by the graph for d = 2. Similar computa-
tional times were observed for the other 4 proteins
with 110-120 amino acids. The computational time
does not include the construction of the α-complex
but it includes the determination of N
∗
d
(i) for each
vertex i. Not surprisingly, the computational time
increases with d as well as with α. However, for
d = 1 and α < 2
˚
A, the computational time is below
2.1 ms. More interestingly, for d = 2 and α < 2
˚
A
(where ε
NV
and ε
NC
are reasonably small), the com-
putational time is below 28 ms. For d = 3 and α < 2
˚
A,
the computational time is below 230 ms. For compar-
ison, computational time with cutoff = 8
˚
A is below
27 ms, with cutoff = 20
˚
A is below 52 ms, and with-
out cutoff is below 216 ms. Hence, picking d = 2 and
α ≈ 1.8
˚
A seems to be a good choice when computing
nonbonded contributions to the potential energies of
protein conformations.
Computational times for 2JZC and 3WCZ are of
course higher. However, for d = 2 and α < 2
˚
A, the
computational time for 2JZC is below 75 ms and it is
below 110-120 ms for 3WCZ.
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
180
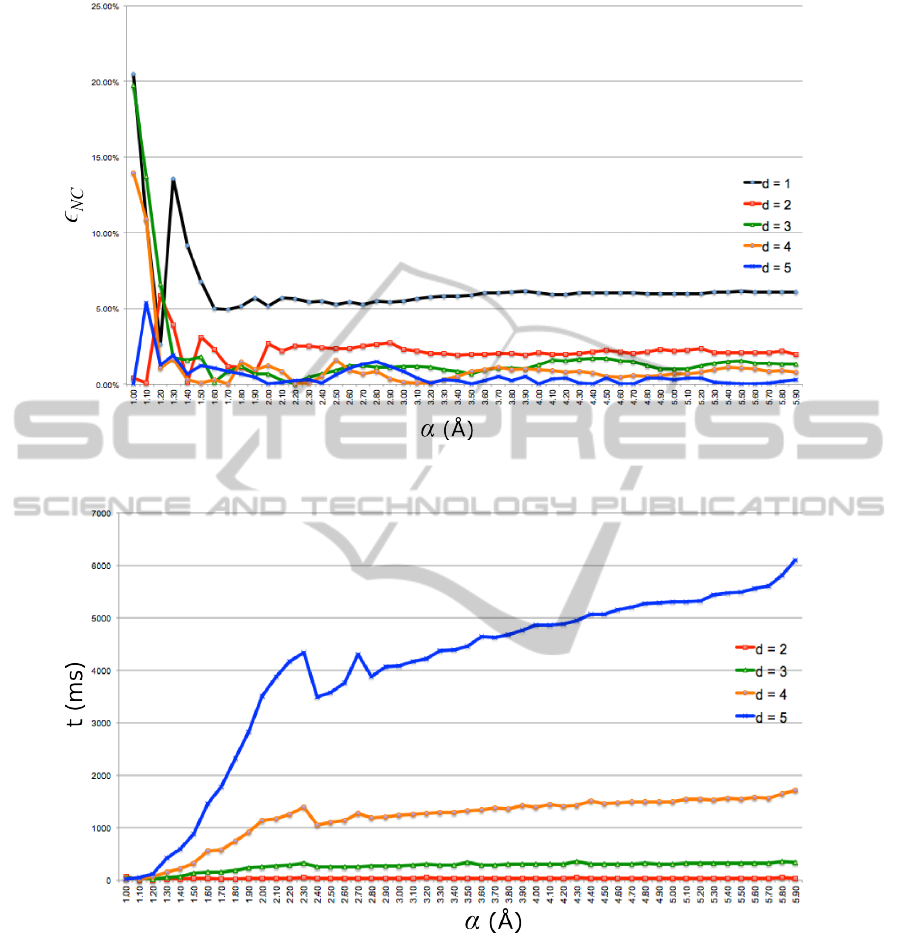
Figure 2: Relative errors for Coulomb interactions in 1X0O.
Figure 3: Computational times for 1X0O (in ms).
5 DISCUSSION
The results show that E
NV
≈ E
∗
NV
and E
NC
≈ E
∗
NC
for
various proteins, already for small d (= 2), and small
α (≈2
˚
A). Hence, 2-complexes seem to provide a use-
ful discrete structure that can be used to speed-up the
determination of the potential energy of protein con-
figurations. This conclusion is of course only valid
if 2-complexes are given beforehand. Otherwise, the
determination of a 2-complexes with 2000 or more
atoms would be computationally much more expen-
sive than when the potential energy is determined by
any cutoff approach.
However, the use of 2-complexes in potential en-
ergy estimations deserves further investigation. In the
protein structure prediction, a vast number of possi-
ble protein configurations is examined when search-
ing for the native one minimizing the potential energy.
The search typically involves moving from one con-
figuration to the next. One way to define neighbor-
AlphaComplexesinProteinStructurePrediction
181

hood of a configuration is by dihedral rotations of one
or several covalent bonds by some, usually small, an-
gle. Atoms on one side of the rotating bond remain
stationary while the others rotate on orbits in parallel
planes and with centers on a common axis. Similarly,
if a covalent bond on a side chain is rotated, only a
very limited number of atoms on the side chain ro-
tates while all other atoms remain stationary.
Kinetic data structures for objects moving on
piecewise continuous trajectories are far from trivial.
The determination of how and when such data struc-
tures must be updated typically involves finding roots
of high-degree polynomials. For DC s, deciding when
a k-simplex T , k ≤ 3, becemes (seizes to be) Delau-
nay involves finding roots of polynomials of 8-th de-
gree (Russel, 2007). In α-complexes, it is necessary
to determine when a k-simplex T , k ≤ 3, becomes
(seizes to be) Gabriel and when it becomes (seizes to
be) short (Kerber and Edelsbrunner, 2013).
Fortunately, when rotating covalent bonds of pro-
teins, the computational effort of updating kinetic
data structures can be significantly reduced. It can
be shown that kinetic DC s and kinetic α-complexes
for this kind of restricted and coordinated movement
of objects (with the same rotational velocity) involve
finding roots of polynomials of degree at most 4. Fur-
thermore, as the results presented in this paper indi-
cate, the depth d of a neighborhood N
∗
d
(i) of vertex i
does not need to be greater than 2 or 3. Hence, these
neighborhoods can be updated efficiently along with
the α-complexes.
In conclusion, α-complexes of proteins with rel-
atively low α do capture the potential energy contri-
butions of nonbonded atoms. Furthermore, kinetic α-
complexes for restricted types of motion can prove
useful in protein structure prediction when searching
through the vast atomic configuration space.
REFERENCES
Edelsbrunner, H. (1992). Weighted alpha shapes. Univer-
sity of Illinois at Urbana-Champaign, Department of
Computer Science.
Edelsbrunner, H., Facello, M., and Liang, J. (1998). On the
definition and the construction of pockets in macro-
molecules. Discrete Applied Mathematics, 88(1):83–
102.
Edelsbrunner, H. and M
¨
ucke, E. P. (1994). Three-
dimensional alpha shapes. ACM Transactions on
Graphics, 13(1):43–72.
Kerber, M. and Edelsbrunner, H. (2013). 3d kinetic al-
pha complexes and their implementation. In ALENEX,
pages 70–77. SIAM.
Kim, D.-S., Seo, J., Kim, D., Ryu, J., and Cho, C.-H.
(2006). Three-dimensional beta shapes. Computer-
Aided Design, 38(11):1179–1191.
Levitt, M., Hirshberg, M., Sharon, R., and Daggett, V.
(1995). Potential energy function and parameters for
simulations of the molecular dynamics of proteins and
nucleic acids in solution. Computer Physics Commu-
nications, 91(1):215–231.
Lotan, I., Schwarzer, F., Halperin, D., and Latombe, J.-
C. (2004). Algorithm and data structures for effi-
cient energy maintenance during monte carlo simula-
tion of proteins. Journal of Computational Biology,
11(5):902–932.
Russel, D. (2007). Kinetic Data Structures in Practice. PhD
thesis, Stanford, CA, USA.
Schlick, T. (2010). Molecular Modeling and Simulation:
An Interdisciplinary Guide, volume 21. Springer.
Seidel, R. (1995). The upper bound theorem for polytopes:
an easy proof of its asymptotic version. Computa-
tional Geometry, 5(2):115–116.
Winter, P. and Fonseca, R. (2012). Adjustable chain
trees for proteins. Journal of Computational Biology,
19(1):83–99.
Winter, P., Sterner, H., and Sterner, P. (2009). Alpha shapes
and proteins. In ISVD’09. Sixth International Sympo-
sium on Voronoi Diagrams, pages 217–224. IEEE.
Zhou, W. and Yan, H. (2014). Alpha shape and delaunay
triangulation in studies of protein-related interactions.
Briefings in Bioinformatics, 15(1):54–64.
BIOINFORMATICS2015-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
182
