
Flow Index based Characterization of next Generation Sequencing
Errors
Visualizing Pyrosequencing and Semiconductor Sequencing to Cope
with Homopolymer Errors
Peter Sarkozy
1
, Márton Enyedi
2
and Peter Antal
1
1
Department of Measurement and Information Systems, Budapest University of Technology and Economics,
Magyar tudósok körútja 2, Budapest, Hungary
2
Institute of Genetics, Biological Research Centre, Hungarian Academy of Sciences, Szeged, Hungary
Keywords: Next Generation Sequencing, Homopolymer Errors, Pyrosequencing, Semiconductor Sequencing,
Visualization.
Abstract: We characterized the error sources of multiple resequencing measurements performed on the Ion Torrent
Personal Genome Machines and the Roche 454 sequencing platforms. Homopolymer insertions and
deletions are the most common error types for these platforms, and there are many underlying factors which
define their occurrence patterns. In the paper we investigate the effect of flow order, specifically the
difference in the average value of the flow values for each homopolymer run length, based on the position in
the flow cycle.
1 INTRODUCTION
Next generation sequencing (NGS) is rapidly
becoming a mature technology. With an increasing
number of platforms distributed throughout the
world, more and more researchers are gaining access
to the world of low-cost, high-throughput
sequencing. The total cost of sequencing is dropping
so rapidly, that other, previously marginal costs like
data analysis are overtaking the cost of consumables.
Researchers must familiarize themselves with the
error characteristics of their chosen platform, or
resort to very stringent quality filtering in order to
identify relevant results amidst the increasing
amount of data.
The first truly NGS platform was the Roche 454
sequencing platform. This technology utilizes clonal
amplification of template libraries on magnetic
beads in emulsion PCR. The beads are then loaded
onto a PicoTiterPlate, where a single bead fits into a
single well. After the addition of reagents
(polymerase, luciferase) to the plate –which allow
the detection of light emitted by luciferase on
nucleotide incorporation–, a repeated order of four
nucleotides are successively flowed over the plate –
with stringent washes in between each flow – and
the light signal generated by the incorporated
nucleotides are recorded and analysed to produce the
base called sequence corresponding the well. Only
one type of nucleotide (A, C, G or T) is flowed in
each cycle, and a nucleotide is incorporated into the
strands only if their complementary nucleotide is the
next free base on the template strand. If multiple
identical bases are next in the template, then the
recorded light signal is proportional to the number of
incorporated bases. Runs of identical nucleotides are
called homopolymers.
Ion Torrent semiconductor sequencing by Life
Technologies uses a very similar approach
(Rothberg et al., 2011). The library preparation stage
also employs emulsion PCR, and coated beads (ion
sphere particles) provide the immobilization of
template strands on a semiconductor plate (chip), but
the detection of nucleotide incorporation is not done
by detecting the light emitted by the luciferase
enzyme, but by using a CMOS semiconductor layer
at the bottom of the plate to detect the change in the
pH of the reaction solution caused by the emission
of a proton when a nucleotide is incorporated into
template strand.
In this paper, we investigate the effect of the
flow index (the position in the flow cycle) on the
271
Sarkozy P., Enyedi M. and Antal P..
Flow Index based Characterization of next Generation Sequencing Errors - Visualizing Pyrosequencing and Semiconductor Sequencing to Cope with
Homopolymer Errors.
DOI: 10.5220/0004924902710277
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2014), pages 271-277
ISBN: 978-989-758-012-3
Copyright
c
2014 SCITEPRESS (Science and Technology Publications, Lda.)

flow values and resulting base calls and error types
in a sequencing run.
2 NGS SEQUENCING ERRORS
There are multiple sources of errors in
pyrosequencing and semiconductor sequencing, and
many of them are common to the two platforms
because of the technological similarity (Quail et al.,
2012, Metzker, 2010).
One of the main sources of errors are the carry
forward/incomplete extension (CAFIE) errors
(Margulies et al., 2005). The carry forward
phenomenon refers to the event when the
nucleotides from a previous flow are not fully
washed out of a well, and these residual nucleotides
incorporate into the clonal template strands if they
match the next complementary nucleotide in the
template.
Carry forward has two major effects: (1) a flow
signal that is higher (since more incorporation events
occurred) than what would be recorded in the
absence of any residual nucleotides in a well, and
(2), the clonal template strands in which the residual
nucleotides are incorporated become desynchronized
(out of phase) compared to the rest of the strands on
a bead, as they are further along in synthesis than the
majority of the clonal templates, and will thus
contribute to the flow signals in different flows than
the rest.
Incomplete extension occurs when not every
nucleotide on a strand whose next nucleotide is
complementary to the current flow is incorporated,
thus resulting in a lower than expected flow signal,
and templates that are lagging behind in synthesis.
As the sequencing progresses with each
successive flow, CAFIE events result in increasingly
asynchronous clonal template strands on each bead.
This effect is directly observable as the baseline
flow signal increases during the sequencing run, and
it results in homopolymer over and undercalls, as
well as single base mismatches.
Many methods have been proposed to help
mitigate the effect of CAFIE errors; including the
alteration of the flow order to allow clonal template
strands to catch up and synchronize, as well as post-
sequencing mathematical methods to model the
effects of CAFIE. The accumulation of CAFIE
errors on a bead during the sequencing run result in
higher variance of the flow signal, and lead to higher
error rates, which are reflected in the read’s base
quality scores. The methods used for reducing
CAFIE errors are commonly referred to as phase
correction, and are highly platform specific and are
implemented in the signal processing and base
calling pipelines of each vendors’ software
platforms.
2.1 Key Signal Normalization
Both sequencing platforms employ a 4-base TACG
key sequence ligated to the 3’ end of each fragment.
This allows easy identification of wells with
populated beads, as well as providing the
normalization levels for the flow signals of each
read. Incorrect normalization can result in multiple
under or overcalls in an affected read.
2.2 Flow Order Optimization
The Ion Torrent Personal Genome Machine
measurements in this paper were sequenced using a
flow order referred to as the Samba, which is a 32
step sequence of
TACGTACGTCTGAGCATCGATCGATGTACAGC
repeated 15.6 times, for a total of 500 flows.
Compared to the previously used flow order of
TCAG, this flow order has the advantage of
allowing the clonal template strands to
resynchronize to some degree, at the expense of non-
optimal read length. It can be demonstrated that for
random sequences, optimal flow order with respect
to read length is attained through repeating the same
flow order of the 4 possible nucleotides. If, for
example the flow order contains only three bases
(e.g. TACTACTACTAC), then all strands are
elongated to the next G nucleotide. The
3 PREVIOUS WORKS
Besides the standard vendor supplied solutions,
multiple approaches have been published that utilize
the flow values underlying the base calls to increase
the base call accuracy of pyrosequencing and
semiconductor sequencing.
3.1 Flow-space Alignment
A read is intrinsically represented as a series of flow
values, where each flow value is proportional to the
length of the corresponding homopolymer run. The
reference sequence can be transformed into series of
flow values, and the alignment can be performed in
flow space with the Flowgram Alignment Tool
(Vacic et al., 2008), allowing higher mapping
accuracy.
BIOINFORMATICS2014-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
272

3.2 Maximum Likelihood Sequence
Clustering
In an early, non-generative approach, a set of
flowgrams were clustered based on the distance
between a flowgram and a sequence, using a
probabilistic model derived from the alignments to
the parent sequence with an exact Needleman-
Wunsch algorithm that empirically models
sequencing noise, and is applied to amplicon
sequencing. Using maximum likelihood, the quantity
and number of true underlying sequences can be
reliably estimated, especially in metagenomics
studies where similar regions are present with
widely varying coverage with PyroNoise (Quince et
al., 2009).
The length of each homopolymer run can be
more accurately characterized using Bayesian
approaches, as the most probable number of
identical bases given the observed flow values with
the help of PyroBayes (Quinlan et al., 2008)
3.3 Hidden Markov Models
Generative approaches attempt to model the
generative process that produces the flowgrams. The
characteristic homopolymer errors in
pyrosequencing and semiconductor sequencing often
require special care when mapping the reads to a
reference sequence. Hidden Markov Models
(HMMs), specifically the pFam models and
extensions widely used in biological sequence
analysis allowed position specific scoring and
management of indels, e.g. to manage that the gap
open penalty of most aligners is often higher than
that of a substitution.
An extended Hidden Markov Model to emit
values in the flow space was proposed as an
integrated solution, which could cope with an error,
where a homopolymer indel follows a base
substitution. The HMM can be constructed with
parameter estimation from the raw flow values and
from the reference sequence transformed into flow
space, and the read alignments to the reference
sequence can be adjusted by decoding the model
with the Viterbi algorithm (Zeng et al., 2013). This
results in higher specificity and sensitivity in variant
detection.
4 MATERIALS AND METHODS
In this paper, we used a human BRCA1 and BRCA2
exon targeted resequencing run on an Ion Torrent
316 chip. All DNA samples were prepared from
blood samples obtained in the Biological Research
Centre (BRC, Szeged). The amplicons (81 PCR
fragments) were generated from germline blood
DNA and covered all coding sequences of the
BRCA1 and BRCA2 genes. These libraries were
prepared with the commercially available Ion Plus
Fragment Library Kit (Life Technologies) using
custom barcoded adaptor sequences.
Generated libraries were controlled for adaptor
dimers and size range using agarose gel
electrophoresis. Samples were isolated from the gel
using DNA fragment extraction kit (Geneaid).
Fragment library quantification was carried out by
Q-PCR (Kapa Biosystems) followed by emulsion
PCR with the Ion PGM™ Template OT2 200 Kit
using the Ion OneTouch™ 2 System (Life
Technologies). Ion sphere particles (ISP) were
enriched using the E/S module and were sequenced
with an Ion PGM in a 200-bp configuration run
using 316 chip (Life Technologies).
The run had a total number of 2,808,212 reads
and an average read length of 115 (σ = 55). The
mapped mismatch rate and insertion/deletion rates
were 0.92%, 0.66%, and 0.5% respectively. The
mismatch rate is inflated by multiple true SNP’s, but
the number insertions and deletions far outnumber
the true indel count in the reference sequence. The
results obtained from our run were compared to the
results from a publicly available Ion Torrent dataset
(FLO-528).
We also tested a publicly available
Acinectobacter baylyi shotgun sequencing run on
the Roche 454 platform as a comparison from the
CloVR public datasets. The dataset had 250,000
reads with an average read length of 467 (σ = 87).
The 454 sequencing measurements analysed in
our research use a fixed flow order of TACG
repeated (though recent advancements have allowed
variable flow order to resynchronize reads) 200
times, for a total of 800 flows.
In order to obtain a deeper understanding of the
underlying measurement, we did not use any quality
clipping or filtering on any of the datasets beyond
that offered by the Torrent Suite 3.6 software
defaults and by the Roche Signal Processing
application defaults, respectively.
The Torrent Suite software as of version 3.2+
does not support the exporting of the results into a
.sff (standard flowgram format) file with phase-
corrected flow values. Since it only exports the raw
key normalized flow values, we created a software
package that allows the conversion of the phase-
corrected flow values from the unaligned BAM files.
FlowIndexbasedCharacterizationofnextGenerationSequencingErrors-VisualizingPyrosequencingandSemiconductor
SequencingtoCopewithHomopolymerErrors
273
This software has been published at
https://github.com/psarkozy/sffviz.
All mapping computations were performed with
BOWTIE2 2.1.0 (Langmead et al., 2012), with the --
very-sensitive option enabled.
5 RESULTS
Previous works acknowledge the significant effect
of the flow index on flow signal distributions, but
they only visualize the flow signal distribution
histograms based on the frequency of each flow
signal value for each nucleotide (Balzer et al., 2010).
Plotting the flow signal distributions vs. the flow
indices allows greater insight into the characteristics
of pyrosequencing and semiconductor sequencing.
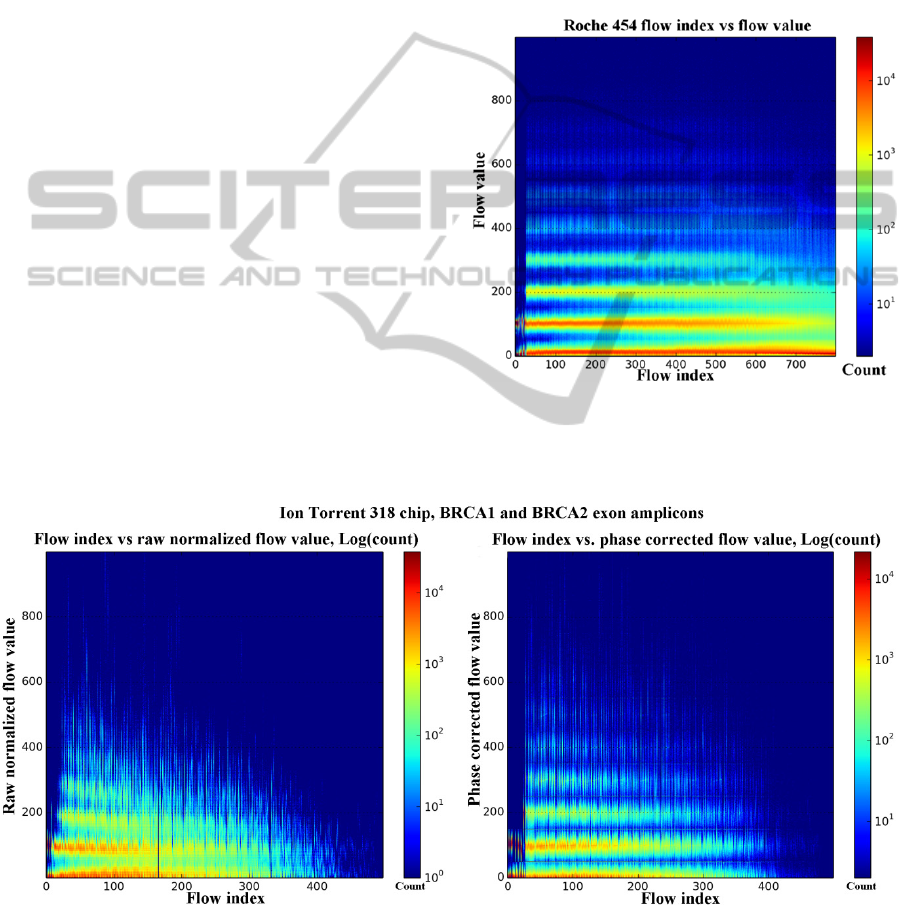
In the case of pyroseqencing, Figure 1. shows
that the spread of the flow signal increases with
homopolymer run length, and also increases with the
flow index. As CAFIE errors accumulate during the
sequencing run, the noise floor of the 0-mer flows
(flows which did not achieve sufficient signal
intensity to classify as a base call) increase, and the
average flow value per mer length decrease. The
flow signal distributions remain easily identifiable
and do not overlap at the beginning of the
sequencing run, but as the flow index increases, they
become more difficult to separate. In
semiconductor sequencing, the raw normalized flow
value histogram (Figure 2.) shows similar
characteristics to pyrosequencing. The variance of
the raw normalized flow values for each
homopolymer run length show greater variance from
the start of the run, and there is a marked signal level
droop as the mer count and flow index increase. A
visible high-frequency jitter is apparent on the
expected flow values of the shorter homopolymer
runs. The noise floor produced also shows a fast
increase rate, especially compared to the 454 dataset
Figure 1: Flow values (Y axis) vs. the flow indices (X
axis) heat map plotted for a Roche 454 shotgun
sequencing run shows good separation of the flow values
corresponding to each homopolymer run length.
Figure 2: The raw normalized (left) and phase corrected (right) flow values from an Ion Torrent 318 chip run. The X axis is
the index of the flow in the flow order, the Y axis represents the flow values. A phase corrected flow value is used to call
the number of bases in a homopolymer run. The cutoff points for each homopolymer length are at multiples of the length.
(100*(mer count)-50). The temperature of the heat map shows the count of each flow index – flow value pair. Flow values
greater than 1000 along with homopolymer lengths of 10+ are clipped from the images to maintain readability.
BIOINFORMATICS2014-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
274
The phase corrected flow values show a more
refined image of the underlying true homopolymer
lengths, and it can be seen that the phase correction
performs well in reducing the signal level droop and
lowering the noise floor. The increased separation of
the flow values corresponding to each mer count is
visible throughout the entire flow sequence. Base
calling is performed on the phase corrected flow
values (Equation 1.).
mer_count = round(flow_value/100) (1)
Single base mismatches occur predominantly in 1-
mer homopolymer base calls, as it is expected based
on the technological platform, with a higher number
of insertions and deletions in the longer
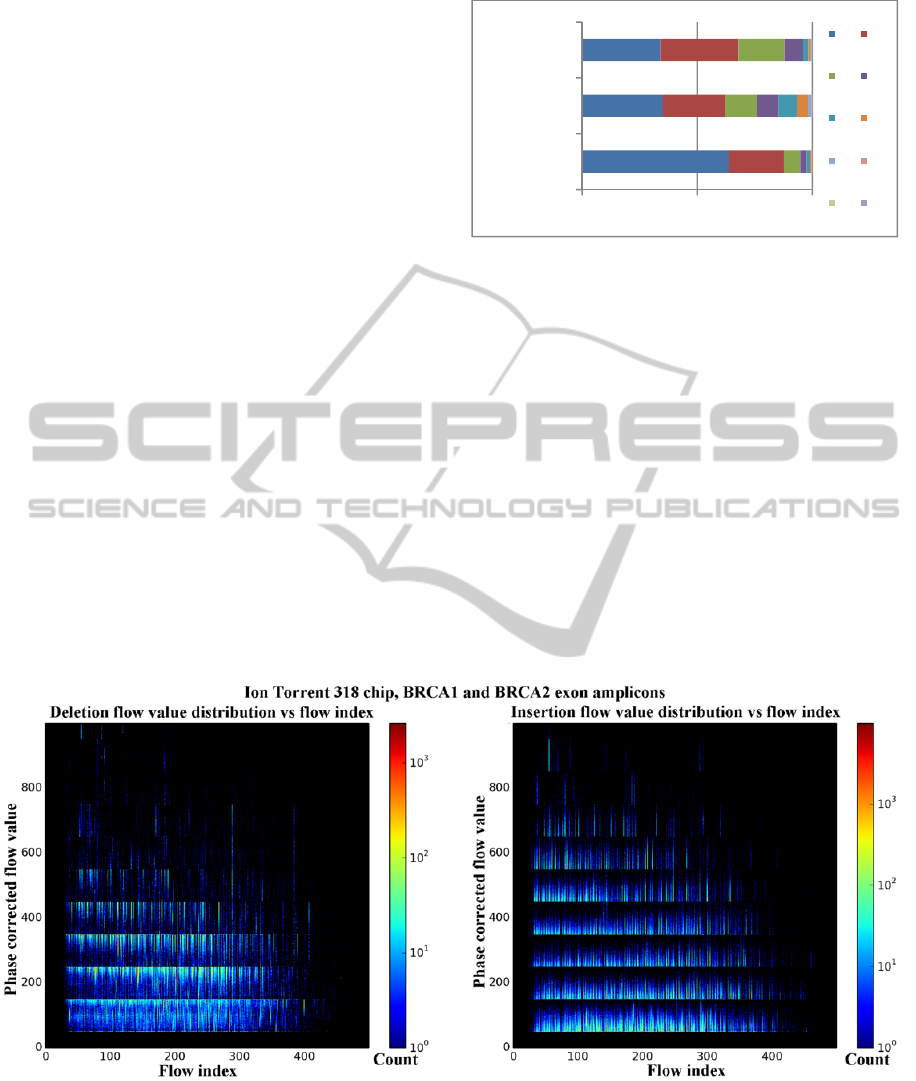
homopolymer runs (Figure 3.). The distribution of
insertions and deletions over the flow index vs. flow
value heat maps (Figure 4.) shows that the values
corresponding to each insertion and deletion usually
occur near the rounding points of Equation 1. The
error rate for all three types of errors increase as the
flow index increases, and the number of errors start
to drop as the read count at each flow index starts to
decrease, as the ends of the reads are surpassed
(Figure 5.).
The high frequency jitter in the flow values
based on the position in the flow cycle has been
observed by others (Bragg et al., 2013). The main
finding presented in the paper is that the cause of
Figure 3: Percentage of errors in N-mer homopolymer
lengths.
this variation in the average flow values per
homopolymer run length can be traced to the
distance between two identical bases in the flow
cycle. Indeed, if the distance between two identical
flows is small (in the Samba cycle, the possible
distance values are characteristic to each base), then
the average flow value corresponding to each mer
count is lower than the expected value of
100*mer_count. Respectively, the average flow
value is higher for greater distances. This
phenomenon is illustrated in Figure 6. Because both
carry forward and incomplete extension errors occur,
the distance metrics should take into account the
distance to the previous identical base, and the next
identical base.
Figure 4: The phase corrected flow value vs. flow index distributions for deletions (left) and insertions (right). The
temperature of the heat map shows the number of indels with a specific flow value and flow index. Homopolymer indels are
clustered near the cutoff points in the flow values, with the majority of deletions closer to the low cutoff points, and
insertions closer to the high cutoff points. The data used for this figure also may contain a small number of non
homopolymer and true indels in the sequencing runs, but their numbers are much lower than the 1% sequencer-specific
error. The final variant calling for this sequencing run is still under evaluation. Flow values greater than 1000 are clipped
from the images for readability.
050100
Mismatch
Insertion
Deletion
1 2
3 4
5 6
7 8
9 10
FlowIndexbasedCharacterizationofnextGenerationSequencingErrors-VisualizingPyrosequencingandSemiconductor
SequencingtoCopewithHomopolymerErrors
275
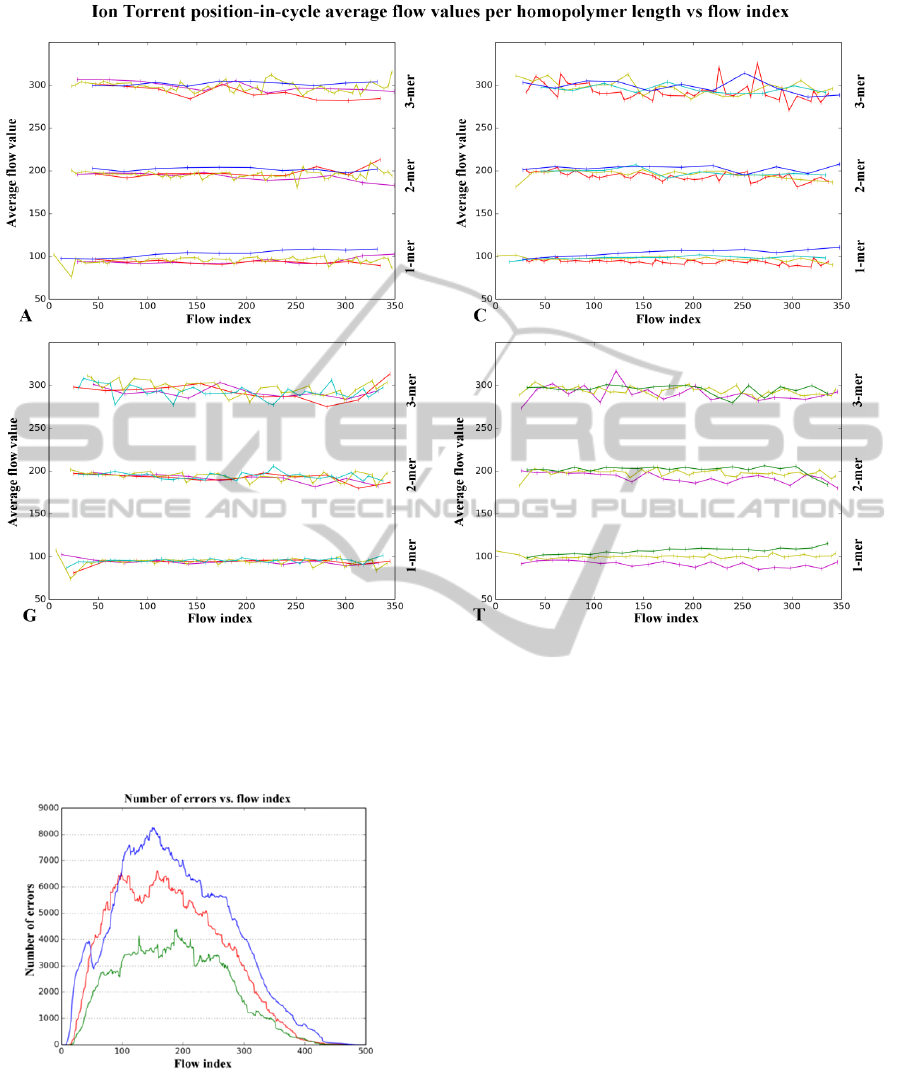
Figure 6: The four bases (A, C, G and T) have different repeat offsets in the Samba flow cycle. Each line represents the
average flow value for a given mer count vs. the flow index. Only mer counts of 1, 2 and 3 are plotted, because low flow
value counts for higher homopolymer lengths result in excessive variance to their average flow values. The lines are color
coded based on the distance of the previous identical base in the flow cycle; magenta = 2, red = 3, yellow = 4, cyan = 5,
green = 6, blue = 7.
Figure 5: The Y axis shows the number of errors in an Ion
Torrent sequencing run against the X axis of the flow
index. The blue line represents base mismatches, the green
line represents deletions, while the red line represents
insertions. The curves are smoothed with a window of 32
(the length of one Samba cycle) to allow visual
differentiation. Note that the counts are not normalized to
the number of reads still under sequencing at each flow
index.
6 CONCLUSIONS
The Samba flow order allows for the mitigation of
the CAFIE errors, but it introduces additional
complexity through dependence on the distance
between identical base flows. In the paper we
investigated this phenomenon, as an explanation for
the difference in the average value of the flow values
for each homopolymer run length, based on the
position in the Samba cycle. The better exploitation
of this effect can lead to improved variant detection
methods. Indeed, current methods are robust for high
coverage sequencing and identifications of germline
mutation, but quantitative applications such as
metagenomics and somatic mutation detection
require higher specificity at lower coverage values.
The software tools developed to convert
unaligned .BAM files exported from the Torrent
Suite software into standard flowgram format file
BIOINFORMATICS2014-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
276

with phase corrected flow values and the
visualization tools are available at
https://github.com/psarkozy/sffviz.
7 FURTHER WORK
The reported findings allow the refinements of
existing generative flowgram based models, to
improve the quality of sequencing measurements.
We are evaluating models that take into account the
position in the flow cycle and the distances to
previous and next identical bases in the flow cycle,
to allow for the correction of the flow signal
distributions, and to enable the reduction of
homopolymer insertions and deletions.
ACKNOWLEDGEMENTS
The publication was supported by the TÁMOP-
4.2.2.C-11/1/KONV-2012-0001 project. The project
has been supported by the European Union, co-
financed by the European Social Fund. This research
was partially supported by the ARTEMIS JU and the
Hungarian National Development Agency (NFÜ) in
frame of the R3-COP (Robust & Safe Mobile Co-
operative Systems) project. The research was also
partially supported by OTKA 81466, OTKA 81941,
OTKA 83766, and GOP-1.1.1-11-2012-0030.
REFERENCES
Rothberg, JM., Hinz, W., Rearick, TM., Schultz, J.,
Mileski, W., 2011. An integrated semiconductor
device enabling non-optical genome sequencing.
Nature 475: 348–352.
Metzker, ML., 2010. Sequencing technologies - the next
generation. Nature Reviews Genetics,11:31-46.
Quail, MA., Smith, M., Coupland, P., Otto, TD., Harris,
SR., Connor, TR., Bertoni, A., Swerdlow, HP., Gu,Y.,
2012. A tale of three next generation sequencing
platforms: comparison of Ion Torrent, Pacific
Biosciences and Illumina MiSeq sequencers. BMC
Genomics 2012. 13:341.
Margulies, M., Egholm, M., Altman, WE., Attiya, S.,
Bader, JS., Bemben, LA., Berka, J., Braverman, MS.,
Chen, YJ., Chen, Z., Dewell, SB., Du, L., Fierro, JM.,
Gomes, XV., Godwin, BC., He, W., Helgesen, S., Ho,
CH., Irzyk, GP., Jando, SC., Alenquer, ML., Jarvie,
TP., Jirage, KB., Kim, JB., Knight, JR., Lanza, JR.,
Leamon, JH., Lefkowitz, SM., Lei, M., Li, J., Lohman,
KL., Lu, H., Makhijani, VB., McDade, KE.,
McKenna, MP., Myers, EW., Nickerson, E., Nobile,
JR., Plant, R., Puc, BP., Ronan, MT., Roth, GT.,
Sarkis, GJ., Simons, JF., Simpson, JW., Srinivasan,
M., Tartaro, KR., Tomasz, A., Vogt, KA., Volkmer,
GA., Wang, SH., Wang, Y., Weiner, MP., Yu, P.,
Begley, RF., Rothberg, JM., 2005. Genome
sequencing in microfabricated high-density picolitre
reactors. Nature. 2005,437:376-80.
Vacic, V., Jin, H., Zhu, JK., Lonardi, S., 2008. A
probabilistic method for small RNA flowgram
matching. Pacific Symposium on Biocomputing
2008:75-86.
Quince, C., Lanzén, A., Curtis, TP., Davenport, RJ., Hall,
N., Head, IM., Read, LF., Sloan, WT., 2009. Accurate
determination of microbial diversity from 454
pyrosequencing data. Nat Methods. 2009,9:639-41.
Quinlan, AR., Stewart, DA., Strömberg, MP., Marth, GT.,
2008. Pyrobayes: an improved base caller for SNP
discovery in pyrosequences. Nature Methods.
2008,5:179 – 18.
Zeng, F., Jiang, R., Chen, T., 2013. PyroHMMsnp: an
SNP caller for Ion Torrent and 454 sequencing data.
Nucleic Acids Research, 2013 Jul;41(13):
Langmead, B., Salzberg, S., 2012. Fast gapped-read
alignment with Bowtie 2. Nature Methods. 2012,
9:357-359.
Balzer, S., Malde, K., Lanzén, A., Sharma, A., Jonassen,
I., 2010. Characteristics of 454 pyrosequencing data—
enabling realistic simulation with flowsim.
Bioinformatics. 2010. 26(18):i420-i425.
Bragg, LM., Stone, G., Butler, MK., Hugenholtz, P.,
Tyson, GW., 2013. Shining a Light on Dark
Sequencing: Characterising Errors in Ion Torrent
PGM Data. PLoS Comput Biol 9(4): e1003031.
FlowIndexbasedCharacterizationofnextGenerationSequencingErrors-VisualizingPyrosequencingandSemiconductor
SequencingtoCopewithHomopolymerErrors
277
