
Generalized Association Rules for Connecting Biological Ontologies
Fernando Benites and Elena Sapozhnikova
Department of Computer and Information Science, University of Konstanz, Konstanz, Germany
Keywords:
Data Mining, Bioinformatics, Generalized Association Rules, Gene Ontology.
Abstract:
The constantly increasing volume and complexity of available biological data requires new methods for man-
aging and analyzing them. An important challenge is the integration of information from different sources
in order to discover possible hidden relations between already known data. In this paper we introduce a data
mining approach which relates biological ontologies by mining generalized association rules connecting their
categories. To select only the most important rules, we propose a new interestingness measure especially
well-suited for hierarchically organized rules. To demonstrate this approach, we applied it to the bioinfor-
matics domain and, more specifically, to the analysis of data from Gene Ontology, Cell type Ontology and
GPCR databases. In this way found association rules connecting two biological ontologies can provide the
user with new knowledge about underlying biological processes. The preliminary results show that produced
rules represent meaningful and quite reliable associations among the ontologies and help infer new knowledge.
1 INTRODUCTION
The constantly increasing volume and complexity of
available biological data calls for new methods of data
management and analysis. The complexity of data
is often caused by the variety of existing interrelated
data sources which all can be used to describe the
same problem. It is especially important in biolog-
ical applications where a single data source can of-
ten reveal only a certain perspective of the underlying
complex biological mechanism. Furthermore, many
single-source-based approaches have been criticized
for their low reliability (Troyanskaya et al., 2003).
In the last years, the bioinformatics community has
encountered the need to integrate information in or-
der to put the data into a useful context, extracting
as much knowledge as possible (Joyce and Palsson,
2006; Carmona-Saez et al., 2006; Hackenberg and
Matthiesen, 2008; Silla and Freitas, 2011).
In this paper we are interested in discovering re-
lationships between categories in complex, hierarchi-
cally structured biological data, particularly ontolo-
gies. Since the use of ontologies facilitates informa-
tion search, storage and understanding, it has been es-
tablished as a standard in biology. Many biomedi-
cal ontologies have been developed for different do-
mains. In the field of genomics the most famous
example is the Gene Ontology (GO). It provides a
structured hierarchy of properties and functional cat-
egories for millions of genes and proteins which are
annotated as belonging to one or more categories (GO
terms). Despite importance of knowledge integration
and the assumption that the most surprising relation-
ships can be found between different domains (Nagel
et al., 2011), the extraction of inter-domain connec-
tions still remains a challenge. To solve this task, data
mining techniques such as association analysis may
help explore dependencies between multiple ontolo-
gies that provide different insights into a certain prob-
lem.
Initially, the association analysis was applied to
the search for sets of elements that frequently co-
occur in a transaction database, i.e. in the market bas-
ket analysis. Co-occurring items build an Association
Rule (AR) of the form X → Y, where X and Y are sets
of items (X is called the antecedent and Y the conse-
quent). In the standard setting, all frequent item sets
are first found by filtering with a minimum support
threshold (support corresponds to the frequency of an
item set in the data). Thereafter, the interestingness of
ARs is measured by confidence which indicates the
estimated conditional probability of a rule given the
elements and the antecedent. And finally, all rules
with the confidence below a user-defined threshold
are pruned. The proper choice of the support and con-
fidence thresholds can become a large problem for the
user because it severely affects the size of the found
rule set.
Generally, AR mining algorithms such as, e.g.,
Apriori (Agrawal et al., 1993) with standard support
229
Benites F. and Sapozhnikova E..
Generalized Association Rules for Connecting Biological Ontologies.
DOI: 10.5220/0004327102290236
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2013), pages 229-236
ISBN: 978-989-8565-35-8
Copyright
c
2013 SCITEPRESS (Science and Technology Publications, Lda.)

and confidence constraints generate a huge amount
of associations, which are largely redundant. This
is an essential drawback for biological applications
(Karpinets et al., 2012; Tseng et al., 2009). De-
spite that confidence was strongly criticized as being
unable to extract truly interesting rules (Brijs et al.,
2003), it is still the most often used Interestingness
Measures (IMs) in bioinformatics.
To improve AR mining, a large number of alter-
native IMs has been proposed in the literature (Lal-
lich et al., 2007). Unfortunately, none of them is
well-suited for mining hierarchically organized data.
In such a case, the redundancy of ARs is caused to
a large extent by the hierarchical structure itself be-
cause rules in higher hierarchy levels subsume rules
in deeper levels. Thus the hierarchy can be success-
fully used for pruning redundant rules by means of
special IMs or hierarchical filtering methods.
The initial idea of hierarchy-based pruning
(Srikant and Agrawal, 1995) is that more special-
ized rules deeper in the hierarchy are pruned un-
less they differ significantly from their ancestor rules.
It was presented as an extension of the standard
support-confidence framework for hierarchical rules
also called Generalized Association Rules (GARs)
with the meaning that they can span different levels
of hierarchies. We will denote it below as General-
ized Rule Pruning (GRP).
We claim that the GAR approach is especially im-
portant for connecting biological ontologies because
the aim of this task is to find the most specialized rules
among interesting ones. The reason is that high-level
rules are often trivial. However for the same reason
the standard support filtering is not appropriate for
this task. As we intend to find all possible connec-
tions between ontologies and not only the most fre-
quent and therefore perhaps the most obvious ones,
we should not use the minimum support threshold.
Developing the discussed ideas, we modified
GRP by replacing support and confidence constraints
through a new hierarchical IM based on the Jaccard
coefficient. We also compared our approach with
GRP and with standard AR mining by several other
IMs. Among them, the first hierarchical IM based on
support and confidence recently proposed by (Benites
and Sapozhnikova, 2012) is especially interesting for
comparison. To illustrate the usefulness of the pro-
posed approach two ground truth datasets were used
at the first step. The number of discovered true as-
sociations was employed as the indicator of the mea-
sure’s quality. Next, the approach was preliminarily
applied to two bioinformatics datasets: GPCR-GO
and CL-GO. The first one is a collection of proteins
from GPCRDB (Vroling et al., 2011) with GO anno-
tations and the second is a collection of articles from
PubMed where terms from Cell Ontology (CL) and
GO co-occurred in sentences.
The rest of the paper is organized as follows: Sec-
tion 2 discusses related work. Section 3 explains our
approach; Section 4 presents the experimental results.
Finally, Section 5 concludes the paper.
2 RELATED WORK
In the last decade, the growth of interest to AR min-
ing can be already seen in bioinformatics, for exam-
ple, in the analysis of micro-array data. Several stud-
ies have been recently conducted to find groups of
co-expressed genes by means of association analy-
sis as an alternative to widely used clustering meth-
ods (Becquet et al., 2002; Creighton and Hanash,
2003; Carmona-Saez et al., 2006; Dafas et al., 2007;
Van Hemert and Baldock, 2007; Tseng et al., 2009;
An et al., 2009). In this context, ARs can describe
relations between expression levels of genes and cer-
tain cellular conditions, for instance, which genes are
over-expressed or under-expressed in diseased cells as
compared to healthy ones.
In (Artamonova et al., 2005; Artamonova et al.,
2007), association analysis was applied to the prob-
lem of finding errors in electronically assigned
functional annotations in large sequence annota-
tion databases. Another interesting application is
the search for predictive combinations of genes in
the genotype-phenotype relationships (Tamura and
D’haeseleer, 2008; MacDonald and Beiko, 2010).
One of the most recent application of association
analysis is presented in (Karpinets et al., 2012) where
classical AR mining is combined with a novel ap-
proach to identify indirect associations and hidden bi-
ological regularities by using semantic-preserving vo-
cabulary and association networks. Two recent works
with the tasks that come close to ours are (Shivakumar
and Porkodi, 2012; Faria et al., 2012). The former ap-
plied the standard Apriori algorithm to connect only
238 GO terms of three GO branches: Molecular Func-
tion (GO-MF), Cellular Component (GO-CC) and bi-
ological process (GO-BP). The same task was previ-
ously solved in (Bodenreider et al., 2005) by three
different approaches: the first one based on similar-
ity in a vector space, the second one based on statisti-
cal analysis of co-occurrence of GO terms, while the
third also dealt with AR mining in the standard set-
ting. In (Faria et al., 2012) the GO-MF annotations
were mined for ARs in order to improve annotation
consistency.
To the best of the authors’ knowledge, all consid-
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
230

ered studies so far are based on the standard AR min-
ing within support-confidence framework and do not
exploit alternative IMs or GARs. We fill this defi-
ciency by using the new IM and modified GRP.
3 METHOD
Our GAR mining approach finds pairwise relation-
ships between categories of multiple ontologies and is
based on (Srikant and Agrawal, 1995). More specifi-
cally, we are given a set of transactions t
i
∈ T which
contains as items categories from multiple ontologies
o
k
i
∈ O
K
classifying the same object. If an object be-
longs to a certain category it should also belong to all
this category’s ancestors. The task is to derive pair-
wise associations between the categories of different
ontologies, i.e. find out how two categories a ∈ X and
b ∈ Y , are related, where X ∩ Y =
/
0, X, Y ⊂ O
K
. Ad-
ditionally, we can solve a similar task which is the
subject of the last experiment where the search for
categories of multiple ontologies is performed in un-
classified data, in particular, in a text corpus. The co-
occurrences of terms corresponding to the categories
of different ontologies in the same sentence enable
building associations between them. A transaction is
therefore represented by a single sentence of the text.
In order to better cope with pruning of GARs, a hi-
erarchical IM Interestingness (Int) based on expected
values for support and confidence as initially pro-
posed by (Srikant and Agrawal, 1995) was recently
introduced in (Benites and Sapozhnikova, 2012). Al-
though it was shown to successfully detect interesting
rules, it has a significant limitation of low noise re-
sistance. It can fail if the expectations become very
small. It happens, for example, if a parent category
has children with highly skewed distributions.
To overcome this problem, we developed a novel
measure Interestingness by Difference (Dif ) which is
based on the difference between the real and the ex-
pected values as follows:
Dif (a, b) = R(a, b)(R(a, b) − E(a, b))
where R is the real value of a given metric for a
rule a → b and E its respectively expectation. Dif de-
pends directly on the magnitude of the real value and
is therefore less sensitive to very small expectations.
If the expected values become greater than the real
ones, Dif converts to negative. This happens, for ex-
ample, when a sibling or even the parent of a node has
a stronger relation to the consequent of the rule.
Let p
a
be the support of a, then to calculate the
expectation, p
ab
is replaced by
p
ˆab
∗p
a
p
ˆa
, i.e., following
the guidelines of GAR but only generalizing on the
antecedent side. We applied Dif to Jaccard coefficient
by deriving the corresponding expectation as follows:
JacExp(a, b) =
p
ˆab
∗p
a
p
ˆa
p
a
+p
b
−
p
ˆab
∗p
a
p
ˆa
, for nodes with parents
and otherwise p
ab
is compared with the case of inde-
pendence, i.e., JacExp(a
r
, b) =
p
a
r
∗p
b
p
a
r
+p
b
−p
a
r
∗p
b
, where
ˆa refers to the parent of a and a
r
to a root node.
The developed IM JacDif was then compared
with the set of measures containing Int, Cosine (Cos)
and All-confidence (ACnf ) from (Surana et al., 2010),
Jaccard (Jac) (Tan et al., 2004), Kulczynski (Kulc)
(Wu et al., 2010), Lift (Lift) (Brin et al., 1997),
Bayes Factor (BF) and Centered Confidence (CCnf )
from (Lallich et al., 2007). We further compared our
method with GRP, assuming that obtained rules are
ranked by confidence.
4 EXPERIMENTS
To examine the proposed approach, four real-world
datasets were used in the experiments. The first two
datasets, called Movies and DBPedia-Yago, had two
ontologies with similar categories and a set of man-
ually created rules connecting them (the so-called
ground truth set). Such an approach is often used
to validate results ((Doan et al., 2002; Maedche and
Staab, 2000)) because it is typically not known how
many and what type of associations should be dis-
covered. In the third and fourth datasets (GPCR-GO
and CL-GO) there were no predefined true rules but
in the last case we used so-called cross-products for
comparison. The cross-products are generated auto-
matically by an information extraction method based
on term decomposition (Bada and Hunter, 2007) and
are then manually verified. Due to pattern match-
ing of substrings in category names, only categories
possessing similar names can be associated by the
method. This is a serious drawback because most
of such connections are trivial ones like “T cell”→”T
cell receptor complex” and therefore not interesting.
Since our method is explicitly designed to avoid dis-
covering obvious rules with respect to the hierarchy,
the comparison with cross-products cannot serve as
the only criterion of its quality.
4.1 Data
The Movies dataset used in (Martin et al., 2008) was
kindly donated to us by Trevor Martin and comprises
movies from the Internet Movie Database (IMDb) and
Rotten Tomatoes (RT) database. We used 3,089 en-
tries which had title and director in both datasets. The
number of categories in the tree-like hierarchies was
GeneralizedAssociationRulesforConnectingBiologicalOntologies
231

88 for IMDb and 76 for RT. To prepare a ground truth
set of associations, 48 connections between the IMDb
and RT categories were created manually: e.g. “Sci-
Fi→Science Fiction and Fantasy”. Due to the small
size of the dataset it is well-suited for the comparison
of IMs.
We further used a large dataset from (Paulheim
and Fümkranz, 2012): DBPedia-Yago. It is based
on the entries of DBPedia which were also tagged by
Yago’s labels. A partial gold standard mapping be-
tween DBPedia and Yago ontologies with 153 links
1
was used as a ground truth set. Our dataset had 271
and 97,680 labels for DBPedia and Yago, respec-
tively. The total number of instances was 159,889.
The third dataset GPCR-GO contained G
protein-coupled receptor proteins from the GPCRDB
database with a tree-like hierarchy. Each of the
proteins was looked up on the UniProtKB on Aug.
2011 to check which GO terms were assigned to
it (excluding annotated electronically (IEA)). For
each of three GO branches (GO-BP, GO-CC, and
GO-MF) the number of removed proteins was differ-
ent, creating different subsets called GPCR-GO-BP,
GPCR-GO-CC and GPCR-GO-MF.
The fourth dataset was introduced by (Hoehndorf
et al., 2008) in order to connect the Cell type Ontol-
ogy (CL) and GO by the text analysis of Pubmed ar-
ticles. Unfortunately, with the data made available
on the Internet we could not reproduce the reported
results and therefore tried to generate the same raw
data. To this end, we downloaded the 56,280 articles
and used the same translation from synsets to spe-
cific terms of ontologies as used by Hoehndorf et al.
We took only the most specific term in a sentence,
removed additional information like references and
glossary and expanded terms with their respective an-
cestors. It resulted in 175,690 sentences with 635 and
6,334 possible terms from CL and GO, respectively.
As we could not find any CL-GO-MF cross-products,
only the cross-products between CL and GO-BP as
well as GO-CC were taken from the Open Biological
and Biomedical Ontologies (OBO) foundry
2
. In to-
tal 196 relationships (from the original 677) actually
co-occurred in the dataset.
To convert each one of the Directed Acyclic Graph
(DAG) hierarchies (DBPedia, Yago, and GO-. . .) into
a tree, we created for every node with multiple par-
ents a new node for each parent, copying the descen-
dants and assuring that each node had only one parent.
However the hierarchies of CL-GO were too deep and
populated, so in this case multiple expectations were
1
http://www.netestate.de/De/Loesungen/DBpedia-
YAGO-Ontology-Matching
2
http://www.obofoundry.org/index.cgi?show=mappings
calculated for multiple parents and the smallest one
was used.
4.2 Finding True Connections
In the first experiment, the impact of using differ-
ent IMs on discovering the true associations of the
Movies and DBPedia-Yago datasets was studied ex-
tensively. For evaluation of results, the well-known
F-1 performance measure which is the harmonic
mean of precision and recall was utilized.
The upper part of Table 1 shows the number of
true rules among the best 48 rules as ranked by each
measure for the Movies dataset. The number of rules
was chosen equal to the total number of true rules.
One can see that JacDif ranked the rules in the best
way as compared to the other measures because it had
the largest number of found true rules (24 or 50%)
followed by BF and Cos. It was though not surprising
that only about a half of all true rules was found. The
reason was that manually created associations were
difficult to discover. So, only eight of them had sup-
port greater than 5%. The second part of Table 1
shows the best possible number of ARs in respect to
F-1 along with the number of true rules found among
them. These values were obtained by the repetitive
rejection of the lowest ranked rule of the set at a time
starting from the whole rule set and by measuring the
corresponding F-1 performance of the restricted rule
set. In this case JacDif again had the best rule set and
also the smallest one.
For the best 48 rules, a direct comparison with Jac,
which found seven true rules less shows that there is
an actual difference in the rule selection between both
measures. In order to analyze it in more depth, we ex-
amined the rules extracted by Jac and compared them
to those extracted by JacDif . There were only one
rule which Jac found but JacDif did not. The rea-
son was that it had a high expectation. On the other
hand, eight true rules were not found by Jac but dis-
covered by JacDif because they had low expectations
and thus high differences between real and expected
values, i.e. were relatively unexpected.
Int had an average score on both, the best 48 and
best possible rule sets. GRP could improve the result
of Cnf but compared against the other metrics it had
a low F-1 value in both cases. Moreover it had the
largest best possible rule set.
Table 2 contains the results of mining DBPedia-
Yago dataset, which are similar to those of Table
1. From the first 153 rules, JacDif found one true
rule less than Jac, but it showed the best F-1 per-
formance on the best possible rule set. This can be
explained by the proper behavior of JacDif which
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
232

Table 1: Movies: The number of found rules and the number of true rules among them (T-rules), for the first 48 rules and for
the best possible rule set. F-1 is in %. Three best values are shown in bold.
Metric Cnf Jac Cos ACnf Kulc Lift BF CCnf GRP Int JacDif
Best 48
T-Rules 7 17 19 16 15 18 21 16 9 15 24
F-1 14.58 35.42 39.58 33.33 31.25 37.50 43.75 33.33 18.75 31.25 50
Best possible
Rules 61 59 58 80 41 44 47 59 165 35 35
T-Rules 9 22 22 26 15 18 21 21 28 14 22
F-1 16.51 41.12 41.51 40.62 33.71 39.13 44.21 39.25 26.29 33.73 53.01
Table 2: DBPedia-Yago: The number of found rules and the number of true rules among them (T-rules), for the first 153 and
for best possible. F-1 is in %. Three best values are shown in bold.
Metric Cnf Jac Cos ACnf Kulc Lift BF CCnf GRP Int JacDif
Best 153
T-Rules 6 75 74 73 73 5 6 29 24 1 74
F-1 3.92 49.02 48.37 47.71 47.71 3.27 3.92 18.95 15.69 0.65 48.37
Best possible
Rules 608 225 201 208 191 260 447 384 581 240 194
T-Rules 105 96 89 92 86 10 33 91 100 3 90
F-1 27.60 50.79 50.28 50.97 50 4.84 11.00 33.89 27.25 1.53 51.87
was able to discover more specific rules as com-
pared with other measures. An example was the
rule “SpaceStation”→“Spacestations” discovered by
JacDif . This choice seems to be quite reasonable
because the parent of “SpaceStation” was “MeanOf-
Transportation” and a connection from it to “Spaces-
tations” would be too general.
Int had the worst F-1 values although on the first
153 rules Cnf , Lift and BF had also very low F-1 val-
ues. The pruning done by GRP could again improve
the result of Cnf for the first 153 rules (since they have
basically the same rule ranking), but it had a slightly
lower result on the best possible set and overall it had
relatively low F-1 values.
After the analysis of the obtained results we se-
lected three measures JacDif , Jac, and ACnf as more
promising for the next experiments. Although the Cos
had slightly better results than ACnf , we chose ACnf
over Cos since it is a more intuitive measure.
4.3 GPCR-GO
In this experiment, there was no ground truth rule set
to be discovered. In our preliminary analysis we fo-
cused mostly on the best 200 rules as ranked by IM
values. The associations between GPCR and GO-MF
were examined in more detail than those of the other
branches.
Although both ontologies focus on proteins and
are therefore similar, the greatest obstacle in connect-
ing them is that their structures are very different. For
example, GPCR and GO have several entries related
to hormones, but whereas GPCR always connect the
term to a protein (group), like “Hormone protein” or
“Gonadotropin-releasing hormone”, GO have more
abstract terms like: “regulation of hormone levels”,
“juvenile hormone secretion”, “hormone transport”,
etc.
Another relevant difficulty was represented by
several missing, inconsistent GO-term assignments or
terms which were too broad
3
This led to a number of
trivial rules discovered by our approach. One example
of such a rule would be GPCR:“Interleukin-8”→GO-
MF:“interleukin-8 binding”. It had the highest JacDif
value of 0.99. However, it should be noted that this
rule can also be found by other methods, especially
by those employing pattern matching on the names,
as can be seen in (Bada and Hunter, 2007).
Another evident rule was GPCR:
“Serotonin”→GO-MF:“serotonin receptor activ-
ity”. But it did not cover all proteins that were
tagged as “Serotonin”, there were three proteins not
connected to the GO-MF term “serotonin receptor
activity”: 5HT6R_HUMAN, Q9W3V5_DROME
and Q9VEG1_DROME. We suggest that it is prob-
ably a missing annotation in GO-MF. It is clear for
5HT6R_HUMAN since this protein is also known as
Serotonin receptor 6. In such a way, an AR can serve
as a start point for investigating the reason why a
3
This inconsistency is caused primarily by varying GO
knowledge of experts and by the fact that not all genes were
tested for each possible GO-term (Faria et al., 2012).
GeneralizedAssociationRulesforConnectingBiologicalOntologies
233

strong rule do not cover all antecedent instances. The
fact that often points to an annotation inconsistency
(Faria et al., 2012). Thus, our approach would assist
GO curators in assignment of missing GO terms to
the GPCR proteins. This research direction is very
important (Artamonova et al., 2005; Artamonova
et al., 2007) and could be a subject of future work.
We could indeed predict several correct GO an-
notations. The rule GPCR:“Chemokine receptor-
like”→GO-MF:“steroid hormone receptor activity”
was ranked much higher by JacDif (rank 33) in com-
parison with Jac (61) and ACnf (66). This rule could
be verified as follows: There were two proteins which
support the rule: GPER_RAT and B3G515_DANRE.
In total, there were three items assigned to
GPCR:“Chemokine receptor-like”, the one missing
was GPER (also known as Q63ZY2_HUMAN). This
last protein was not annotated by the term GO-
MF:“steroid hormone receptor activity” at the time
the data were gathered nor had any GO term manu-
ally curated. The only GO-MF term assigned to it
at this time was GO-MF:“G-protein coupled recep-
tor activity” and it was an IEA term. This year it
obtained the assignment to GO-MF:“steroid hormone
receptor activity”
4
by Ensemble Compara
5
based on
electronic inference from the GPER_RAT annota-
tion. Our method could also lead to such an IEA
without any additional information. Another inter-
esting rule found by our approach was GPCR:“Beta
Adrenoceptors”→GO-MF:“protein dimerization ac-
tivity”. It was ranked 74th, 78th, and 80th by JacDif ,
Jac, and ACnf , respectively. There were 32 proteins
classified as GPCR:“Beta Adrenoceptors”, and only
28 of them corresponded to this rule. One of four
proteins not assigned to the GO-MF:“protein dimer-
ization activity” was D4ACM3_RAT that obtained the
annotation of GO-MF:“protein homodimerization ac-
tivity” in February 2011 in the RGD
6
. This term is
a direct child of GO-MF:“protein dimerization activ-
ity” and it has not been assigned in UniProtKB until
now
7
.
JacDif generally ranked rules higher if they
could be seen as more surprising from the per-
spective of the hierarchy. This can be illustrated
by the rule GPCR:“Muscarinic acetylcholine”→GO-
MF:“G-protein coupled acetylcholine receptor activ-
ity” ranked 7th as compared with the aforementioned
serotonin rule, which was ranked by JacDif only 14th
(by ACnf 12th and by Jac 9th). The former rule was
4
Stand of Aug. 8th 2012
5
http://www.ensembl.org/info/docs/api/compara/
index.html
6
The Rat Genome Database: http://rgd.mcw.edu
7
Stand of Aug. 8th 2012
ranked higher because its expectation was equal to
0.04 whereas the actual Jac value was 0.93. The sero-
tonin rule, in turn, had the same Jac value, but its ex-
pectation was much higher 0.13.
The first examination of obtained connections in
the ontology pairs GPCR-GO-BP and GPCR-GO-CC
showed that their analysis requires more investiga-
tions because the more complex GARs in these cases
can actually help infer new knowledge. We were able
to verify the known facts that C-C Chemokine type
2 is connected to the regulation of T-cell prolifera-
tion (Schjetne et al., 2003), whereas type 7 is con-
nected to the regulation of hypersensitivity (Schneider
et al., 2007). A possible missing entry could be found
by the analysis of the rule GPCR:“Serotonin type
2b”→GO-BP:“regulation of autophagy”. Although
the protein 5HT2B_HUMAN supports the rule, the
protein 5HT2B_TETFL (serotonin type 2b from the
tetraodon) was not assigned to it. Other rules need
further analysis and can probably lead to the discov-
ery of additional interesting cases.
4.4 CL-GO
As stated before, the partial cross-products were used
as a ground truth rule set in this experiment. How-
ever the serious disadvantage of this evaluation is that
the cross-products represent obvious associations (by
connecting only categories with similar names) and
can therefore be ranked lower than less obvious rules
by our approach. In Fig. 1 one can see the increase
in the number of found true rules among the best X
rules with growing X. The graph shows that JacDif
could find more true rules than Jac and ACnf among
the same number of extracted rules. The low numbers
of true rules found in this experiment as compared to
those of Movies and DBPedia-Yago point to the fact
that other more interesting and unexpected rules were
ranked higher.
Among the top ranked rules, several in-
teresting connections from CL to GO like
“heterocyst”→“nitrogen fixation” were discov-
ered. Heterocyst is a differentiated cyanobacterial
cell that carries out nitrogen fixation
8
. It is important
to note that such rules could not be found by the
name matching approach of (Bada and Hunter, 2007)
and therefore were absent in the cross-products. Our
approach could also find some associations between
categories with similar names like “nitrogen fixing
cell”→“nitrogen fixation” which were nevertheless
not contained in the cross-products obtained from
the OBO foundry. Other examples of interesting
rules were: “glandular cell of stomach”→“acid
8
http://www.uniprot.org/keywords/364
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
234
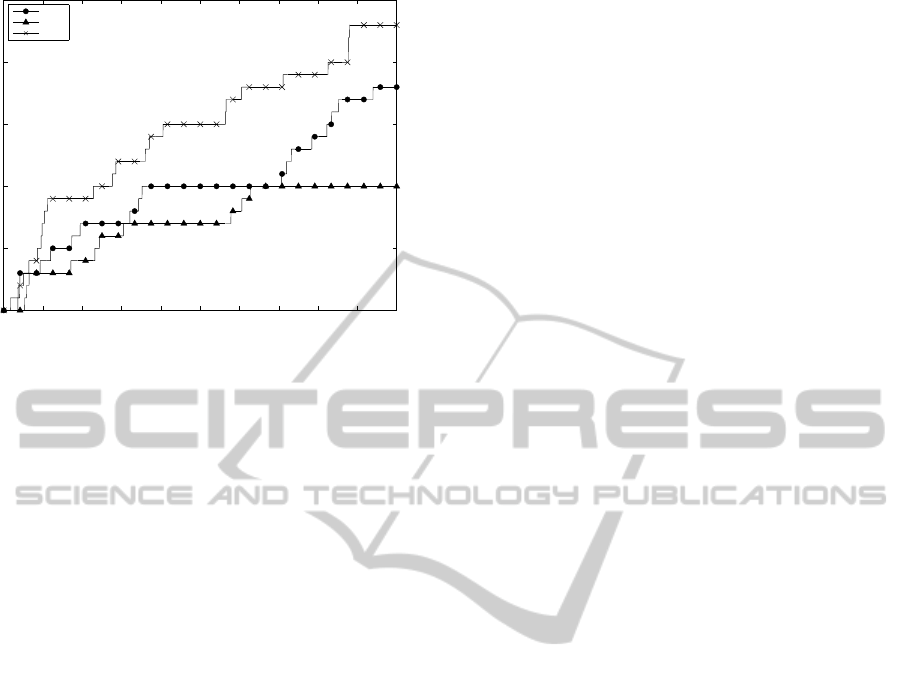
Figure 1: Number of true rules found among the best X
rules by JacDif , Jac, and ACnf for CL-GO.
secretion”, “spermatocyte”→“meiosis I” and
“osteoclast”→“bone remodeling”.
5 CONCLUSIONS
In this paper we have examined connecting multiple
biological ontologies by association analysis. Asso-
ciations found between classes of different ontologies
can be used to support existing knowledge or to ex-
tract new knowledge with the aim of better under-
standing biological mechanisms. We proposed min-
ing Generalized Association Rules (GARs) by means
of a new interestingness measure especially devel-
oped for hierarchically organized rules.
Our approach was applied to four real-world
datasets from the areas of text mining and bioinfor-
matics. The proposed measure was compared with
conventional measures and another hierarchical mea-
sure as well as with the standard GRP on the first two
datasets with the ground truth rule sets. It achieved
the best results in terms of the F-1 performance mea-
sure. In the third and fourth experiments it was able
to extract more interesting and more specific rules
as compared to the best conventional interestingness
measures.
The preliminary analysis of these rules revealed
meaningful associations between certain genes and
proteins which make sense biologically and some
other rules which need further investigations and can
lead probably to the generation of new hypotheses to
explain them. Such investigations are the subject of
our future work. It was also shown that associations
extracted with our method can help GO curators as-
sign missing GO terms by analyzing deviations from
high ranked rules because such deviations are often
caused through inconsistent annotations. Since our
approach utilizes several information sources, it pro-
vides more deep insights into protein annotations than
the methods based only on one source. Thus it can
be integrated in automatic GO term assignment algo-
rithms or used manually for revising ontologies. As
future work, we additionally plan a further application
of the proposed approach to find annotation inconsis-
tencies in the field of protein function prediction.
REFERENCES
Agrawal, R., Imieli
´
nski, T., and Swami, A. (1993). Min-
ing Association Rules between Sets of Items in Large
Databases. In Proc. of the 1993 ACM SIGMOD Int.
Conf. on Management of Data.
An, L., Obradovic, Z., Smith, D., Bodenreider, O., and
Megalooikonomou, V. (2009). Mining association
rules among gene functions in clusters of similar gene
expression maps. In 2nd Wksp. on Data Mining in
Functional Genomics.
Artamonova, I., Frishman, G., and Frishman, D. (2007).
Applying negative rule mining to improve genome an-
notation. BMC Bioinformatics, 8.
Artamonova, I., Frishman, G., Gelfand, M., and Frishman,
D. (2005). Mining sequence annotation databanks for
association patterns. Bioinformatics, 21(3).
Bada, M. and Hunter, L. (2007). Enrichment of obo ontolo-
gies. J. of Biomed. Informatics, 40(3).
Becquet, C., Blachon, S., Jeudy, B., Boulicaut, J., and Gan-
drillon, O. (2002). Strong-association-rule mining
for large-scale gene-expression data analysis: a case
study on human SAGE data. Genome Biol., 3(12).
Benites, F. and Sapozhnikova, E. (2012). Learning Different
Concept Hierarchies and the Relations Between them
from Classified Data. Intel. Data Analysis for Real-
Life Appl.: Theory and Practice.
Bodenreider, O., Aubry, M., and Burgun, A. (2005). Non-
lexical approaches to identifying associative relations
in the gene ontology. In Pacific Symp. on Biocomput-
ing.
Brijs, T., Vanhoof, K., and Wets, G. (2003). Defining in-
terestingness measures for association rules. Int. J. of
Inf. Theories and Appl., 10(4).
Brin, S., Motwani, R., Ullman, J., and Tsur, S. (1997). Dy-
namic itemset counting and implication rules for mar-
ket basket data. In Proc. of the ACM SIGMOD Int.
Conf. on Manag. of data.
Carmona-Saez, P., Chagoyen, M., Rodríguez, A., Trelles,
O., Carazo, J., and Pascual-Montano, A. (2006). Inte-
grated analysis of gene expression by association rules
discovery. BMC Bioinformatics, 7.
Creighton, C. and Hanash, S. (2003). Mining gene expres-
sion databases for association rules. Bioinformatics,
19(1).
Dafas, A., Garcez, D., and Artur, S. (2007). Discovering
Meaningful Rules from Gene Expression Data. Curr.
Bioinformatics, 2(3).
GeneralizedAssociationRulesforConnectingBiologicalOntologies
235

Doan, A., Madhavan, J., Domingos, P., and Halevy, A.
(2002). Learning to map between ontologies on the
semantic web. In Proc. of the 11th Int. Conf. on WWW.
Faria, D., Schlicker, A., Pesquita, C., Bastos, H., Ferreira,
A. E., Albrecht, M., and Falcão, A. (2012). Mining
go annotations for improving annotation consistency.
PLoS ONE, 7.
Hackenberg, M. and Matthiesen, R. (2008). Annotation-
Modules: A tool for finding significant combinations
of multisource annotations for gene lists. Bioinformat-
ics.
Hoehndorf, R., Ngonga, A., Dannemann, M., and Kelso,
J. (2008). From terms to categories: Testing the sig-
nificance of co-occurrences between ontological cat-
egories. In Proc. of the 3rd Int. Symp. on Semantic
Mining in Biomed.
Joyce, A. R. and Palsson, B. O. (2006). The model organism
as a system: integrating ’omics’ data sets. Nat. Rev.
Mol. Cell. Biol., 7(3).
Karpinets, T., Park, B., and Uberbacher, E. (2012). Ana-
lyzing large biological datasets with association net-
works. Nucleic Acids Research.
Lallich, S., Teytaud, O., and Prudhomme, E. (2007). As-
sociation rule interestingness: Measure and statistical
validation. In Quality Measures in Data Mining, Stud-
ies in Comp. Intel.
MacDonald, N. and Beiko, R. (2010). Efficient learning
of microbial genotype-phenotype association rules.
Bioinformatics, 26(15).
Maedche, A. and Staab, S. (2000). Discovering conceptual
relations from text. In Proc. of the 14th ECAI.
Martin, T., Shen, Y., and Azvine, B. (2008). Granular asso-
ciation rules for multiple taxonomies: A mass assign-
ment approach. Uncertainty Reasoning for the Seman-
tic Web I.
Nagel, U., Thiel, K., Kötter, T., Piatek, D., and Berthold, M.
(2011). Bisociative discovery of interesting relations
between domains. In Proc. of the 10th Int. Symp. on
Intel. Data Analysis, Lecture Notes in Computer Sci-
ence (LNCS).
Paulheim, H. and Fümkranz, J. (2012). Unsupervised gen-
eration of data mining features from linked open data.
In Proc. of the 2nd Int. Conf. on Web Intel., Mining
and Semantics.
Schjetne, K., Gundersen, H., Iversen, J.-G., Thompson, K.,
and Bogen, B. (2003). Antibody-mediated delivery of
antigen to chemokine receptors on antigen-presenting
cells results in enhanced cd4+ t cell responses. Euro-
pean J. of Immunology, 33(11).
Schneider, M., Meingassner, J., Lipp, M., Moore, H., and
Rot, A. (2007). Ccr7 is required for the in vivo func-
tion of cd4+ cd25+ regulatory t cells. The J. of Exp.
Med., 204(4).
Shivakumar, B. and Porkodi, R. (2012). Finding relation-
ships among gene ontology terms in biological doc-
uments using association rule mining and go annota-
tions. Int. J. of Computer Science, Inf. Tech., & Secu-
rity, 2(3).
Silla, C. and Freitas, A. (2011). Selecting different protein
representations and classification algorithms in hierar-
chical protein function prediction. Intel. Data Analy-
sis, 15(6).
Srikant, R. and Agrawal, R. (1995). Mining generalized
association rules. In Proc. of the 21th Int. Conf. on
Very Large Data Bases.
Surana, A., Kiran, U., and Reddy, P. (2010). Selecting
a right interestingness measure for rare association
rules. In 16th Int. Conf. on Manag. of Data.
Tamura, M. and D’haeseleer, P. (2008). Microbial
genotype-phenotype mapping by class association
rule mining. Bioinformatics, 24(13).
Tan, P., Kumar, V., and Srivastava, J. (2004). Selecting the
right objective measure for association analysis. In-
formation Systems, 29.
Troyanskaya, O., Dolinski, K., Owen, A., Altman, R., and
Botstein, D. (2003). A Bayesian framework for com-
bining heterogeneous data sources for gene function
prediction (in S. cerevisiae).
Tseng, V., Yu, H., and Yang, S. (2009). Efficient mining of
multilevel gene association rules from microarray and
gene ontology. Inform. Syst. Front.
Van Hemert, J. and Baldock, R. (2007). Mining spatial gene
expression data for association rules. In Proc. of the
1st int. conf. on Bioinformatics research and develop-
ment, BIRD’07.
Vroling, B., Sanders, M., Baakman, C., Borrmann, A., Ver-
hoeven, S., Klomp, J., Oliveira, L., de Vlieg, J., and
Vriend, G. (2011). Gpcrdb: information system for
g protein-coupled receptors. Nucleic Acids Research,
39(suppl 1).
Wu, T., Chen, Y., and Han, J. (2010). Re-examination of
interestingness measures in pattern mining: a unified
framework. Data Min. Knowl. Disc., 21.
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
236
