
Structural Analysis of Nuclear Magnetic Resonance Spectroscopy
Data
Alejandro Chinea
1
and José L. González-Mora
2
1
Departamento de Física Fundamental, Facultad de Ciencias UNED, Paseo Senda del Rey nº9, 28040, Madrid, Spain
2
Departamento de Fisiología, Facultad de Medicina ULL, Campus de Ciencias de la Salud, 38071, La Laguna, Spain
Keywords: NMR Spectroscopy, Clinical Diagnosis, Machine Learning Applications.
Abstract: From the clinical diagnosis point of view in vivo nuclear magnetic resonance (NMR) spectroscopy has
proven to be a valuable tool for performing non-invasive quantitative assessments of brain tumour glucose
metabolism. Brain tumours are considered fast-growth tumours because of their high rate of proliferation.
Therefore, there is strong interest from the clinical investigator’s point of view in the development of early
tumour detection techniques. Unfortunately, current diagnosis techniques ignore the dynamic aspects of
these signals. It is largely believed that temporal variations of NMR spectra are simply due to noise or do
not carry enough information to be exploited by any reliable diagnosis procedure. Thus, current diagnosis
procedures are mainly based on empirical observations extracted from single averaged spectra. In this paper,
a machine learning framework for the analysis of NMR spectroscopy signals is introduced. The proposed
framework is characterized by a set of structural parameters that are shown to be very sensitive to metabolic
changes as those exhibited by tumour cells. Furthermore, they are able to cope not only with high-
dimensional characteristics of NMR data but also with the dynamic aspects of these signals.
1 INTRODUCTION
The last decade has seen a rise in the application of
proton NMR spectroscopy techniques,
fundamentally in fields such as biological research
(Raamsdonk et al., 2001) and clinical diagnosis
(Lisboa et al., 2010). The main goal within the
biological research field is to achieve a deep
understanding of metabolic processes that may lead
to advances in many areas including clinical
diagnosis, functional genomics, therapeutics and
toxicology. In addition, metabolic profiles from
proton NMR spectroscopy are inherently complex
and information-rich, thereby having the potential to
provide fundamental insights into the molecular
mechanisms underlying health and disease.
Nevertheless, it is important to note that the main
difficulty is not simply how to extract the
information efficiently and reliably but how to do so
in a way which is interpretable to people with
different technical backgrounds. In fact, machine
learning techniques (Bishop, 1995; 2006) have
recently been recognized by biological researchers
(Ebbels and Cavill, 2009) as an important method
for extracting useful information from empirical
data.
From a clinical diagnosis point of view, proton
NMR spectroscopy has proven to be a valuable tool
which has benefited from the knowledge and
experience acquired through biological research
studies. Furthermore, a large number of proton NMR
spectroscopy applications have targeted the human
brain. Specifically, it has been extensively used for
the study of brain diseases and disorders, including
epilepsy (Aydin et al., 2007), schizophrenia
(Sigmundsson et al., 2003), parkinson's disease
(Summerfield et al., 2002) and bipolar disorder
(Frye et al., 2007) amongst others. This is mainly
due to the fact that proton NMR spectroscopy is a
non-invasive technique, which is particularly
important in this part of the body where clinical
surgery or biopsy is more delicate than in other
areas. In addition, it is important to note that it
allows in vivo quantification of metabolite
concentrations in brain tissue for clinical diagnosis
purposes. Moreover, one of the most successful
applications of proton NMR spectroscopy has been
cancer research (Kwock et al., 2006); (Bottomley,
1984). In this paper, the focus is on proton NMR
brain spectroscopy.
Generally speaking, the process of clinical
diagnosis involves the analysis of spectroscopy
212
Chinea A. and L. González Mora J..
Structural Analysis of Nuclear Magnetic Resonance Spectroscopy Data.
DOI: 10.5220/0004321902120222
In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms (BIOINFORMATICS-2013), pages 212-222
ISBN: 978-989-8565-35-8
Copyright
c
2013 SCITEPRESS (Science and Technology Publications, Lda.)

signals obtained from a well-defined cubic volume
of interest (single voxel experiment) in a specific
region of the brain during a pre-defined time frame
(acquisition time). Two acquisition methods are
commonly used, namely point resolved spectroscopy
(PRESS) (Frahm et al., 1987) or stimulated echo
acquisition mode (STEAM) (Nelson and Brown,
1987). Most of the time, the analysis of the signals is
carried out in the frequency domain. The raw signal
from the free induction decays (FIDs) is transformed
using the discrete fourier transform. Afterwards, a
pre-processing stage is also performed to remove
artefacts from the acquisition process. Finally, the
resulting spectral signals, whose number is
approximately equal to the acquisition time divided
by the repetition time of the sequence, are averaged
and in most cases used for a preliminary diagnosis
which relies on a simple visual analysis of the
spectra.
However, current diagnosis techniques based on
proton NMR spectroscopy are still in their infancy.
Firstly, as stated above, powerful tools like machine
learning techniques are scarcely applied within this
context (Sadja, 2006). Indeed, most of the
applications of machine learning techniques have
been in the field of systems biology research
(Friedman, 2004); (Basso et al., 2005) and have used
data from other techniques like liquid
chromatography mass spectrometry or gene
expression microarray. This is mainly due to the
abovementioned problems regarding the
interpretability of the information but also because
of a lack of effective communication of results
between researchers working in different fields. Of
particular interest is the fact that current diagnosis
techniques ignore the dynamic aspects of these
signals. It is largely believed that the information
content of temporal variations of NMR Spectra is
minimal. Thus, current diagnosis procedures are
constrained to empirical observations extracted from
a single averaged spectrum. Furthermore, this fact
could mask important information concerning
metabolic changes, especially in early stages of
tumour formation. In this paper, a machine learning
framework for the analysis of NMR spectroscopy
signals is introduced which is able to exploit both
static and dynamic aspects of these signals.
The rest of this paper is organized as follows: In
the next section, the principal characteristics and
difficulties associated with the processing of H-
NMR signals are presented. In section 3, a formal
characterization of NMR-based data is introduced
from a machine learning point of view. The
reliability of the proposed measures is assessed
through careful analysis of the results provided by a
specific experimental design in section 4. Finally,
section 5 provides a summary of the present study
and some concluding remarks.
2 CHARACTERISTICS OF
H-NMR DATA
The metabolites detectable with proton NMR
spectroscopy include, between others, the
resonances of N-acetylaspartate (NAA), N-acetyl
aspartyl glutamate (NAAG), alanine (Ala), Choline
(Cho), creatine (Cr), gamma-aminobutyric acid
(GABA), glutamine (Gln), and a variety of other
resonances that might not be evident depending on
the type and quality of spectra as well as on the
pathological condition. The molecular structure of a
particular metabolite is reflected by a typical peak
pattern. Furthermore, the area (amplitude) of a peak
is proportional to the number of nuclei that
contribute to it and therefore to the concentration of
the metabolite to which the nuclei belong.
Of particular interest is the fact that even if peak
amplitudes change from different samples reflecting
a change in concentration, the ratios between the
central resonance peak and sub-peaks, composing
the metabolite fingerprint, always remain constant.
Most of the metabolites have multiple resonances
many of which are split into multiplets as a result of
homonuclear proton scalar coupling. Despite high
magnetic fields increase the sensitivity and spectral
dispersion in NMR spectroscopy, at clinical field
strengths (from 1.5 up to 3 Teslas), there exists a
significant overlap of peaks from different
metabolites.
In addition, the response of coupled spins is
strongly affected by the acquisition parameters of
the NMR sequence, e.g. radio frequency pulses
employed and the time intervals set between them
(Cloarec et al., 2005). Furthermore, additional
difficulties are caused by the presence of
uncharacterized resonances from macromolecules or
lipids. This is further complicated by small but
significant sample to sample variations in the
chemical shift position of signals, produced by
effects such as differences in pH and ionic strength.
As a result of this problem, information coming
from a given metabolite contaminates the spectral
dimensions containing information from other
metabolites. In other words, due to this phenomenon
the resonance frequency of certain metabolites can
suffer slight variations from sample to sample.
StructuralAnalysisofNuclearMagneticResonanceSpectroscopyData
213
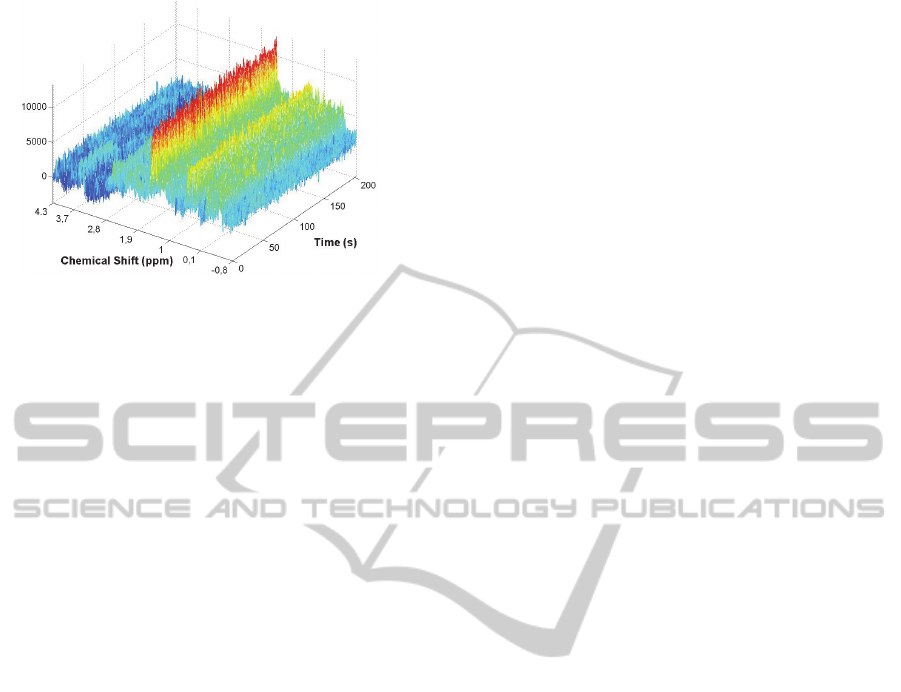
Figure 1: Temporal evolution of the Spectra associated to
a short-TE NMR single voxel brain spectroscopy
experiment at 3T (3 Teslas of Magnetic field strength).
The voxel was located in the visual cortex of a healthy
patient. A total echo time (TE) of 23 ms and a repetition
time (TR) of 1070 ms were used for the acquisition
process conducted during several minutes. The molecular
structure of a particular metabolite is reflected by a typical
peak pattern. The area (amplitude) of a peak (i.e., the
vertical axis of spectra) is proportional to the number of
nuclei that contribute to it and therefore to the
concentration of the metabolite to which the nuclei belong.
The horizontal axes correspond to the chemical shift scale
(ppm) axis (which is representing brain metabolites
resonances) and the time axis (in seconds) respectively.
For instance, the resonance associated to the N-
acetylaspartylglutamate (NAAG), a dipeptide of N
substituted aspartate and glutamate (that is believed to be
involved in excitatory neurotransmission processes) is
located at 2.046 ppm.
In order to address these problems, some
sophisticated strategies have been proposed
(Bruschweiler and Zhang, 2004); (Keun et al.,
2008). However, the common factor to all the
abovementioned techniques is that they completely
ignore the inherent dynamics of NMR spectroscopy
signals. Unfortunately, it is largely believed that
temporal variations of NMR Spectra (see figure 1)
are simply noise or do not carry enough information
to be exploited by any reliable diagnosis procedure.
For example, in a single-voxel MRS experiment, as
a result of the acquisition process, a whole matrix of
data is obtained from the region of interest.
Furthermore, within that matrix two consecutive
rows correspond to signal frames taken with a time
difference equal to the repetition time set for the
acquisition sequence (usually 1035-1070 ms for
short total echo time sequences). Therefore, the
number of rows (signal frames) is approximately
equal to the acquisition time divided by the
repetition time of the sequence. Indeed, there is a
small number of frames that are used for water
referencing (usually eight) which are suppressed
when generating the data matrix. In addition, each
row represents a spectral signal obtained from the
volume of interest after a pre-processing stage which
transforms the raw FID temporal data into the
frequency domain. The frequency domain is usually
preferred over the temporal domain (Vanhamme et
al., 2001) since this enables visual interpretation.
Moreover, in the frequency domain the NMR
signal is represented as a function of resonance
frequency. Additionally, each column represents a
given metabolite or metabolic signal. The number of
columns depends on the particular pre-processing
technique used, but is usually 8192 or 4096
dimensions, depending whether or not a zero filling
procedure is applied to the transformed signal. In
both cases, the associated chemical shift range
corresponds approximately to the interval [-14.8,
24.8] ppm. However, as stated above, the range of
interest usually taken for brain metabolites is
restricted to the interval [-0.8, 4.3 ppm] leading to a
dimensionality reduction with respect to the original
row size. The exact dimension of these vectors
depends on the procedure used for generating the
chemical shift scale. The water peak and the creatine
peak (i.e., one of the 35 known metabolites involved
in brain metabolism) are commonly used for this
purpose. Nevertheless, it is important to note that the
resulting matrix after selecting the appropriate range
is still composed of high-dimensional patterns.
Afterwards, the temporal variations of the spectra
(i.e. the rows of the matrix of data) are averaged in
order to obtain a single vector (single spectrum
signal). In such vectors each dimension represents
the mean value of a particular metabolic signal.
3 STRUCTURAL MEASURES
In the following sub-sections we introduce a set of
measures that have been used within the context of
supervised learning (Haykin, 1999); (Cherkassy and
Mulier, 2007) for the structural characterization of
NMR-based data sets. The principal advantage of
these characterization parameters is not only their
simplicity but also the fact that they do not make any
assumption about the underlying nature of the data.
Therefore, they are appropriate for dealing with both
static and dynamic data sets.
Without a loss of generality let us suppose a data
set
N
D composed of N patterns belonging to a
space of dimension d. Furthermore, it is assumed
that each sample of the data set belongs to a
category
i
w where i = 1,2,….,C. In other words,
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
214

there are C different pattern categories (or classes)
defined in the input space. If we group the input
variables
i
x into a vector
d
xxxxx ,...,,,
321
,
the data set can be formally defined as a set of
vectors
k
x
in d dimensions (i.e., patterns) where
Nk 1 , where each pattern belongs to one of
the categories defined in the input space,
N
k
Dx
the category of a pattern
k
x
is
represented using the notation
i
k
wxclass
where
Ci 1 .
3.1 Inertia
Inertia (Blayo et al., 1995) is a classical measure for
the variance of high dimensional data. We
distinguish here three types of inertia, namely,
global inertia, within-category inertia and between-
category inertia:
N
k
k
G
x
N
I
1
2
1
(1)
i
kk
N
k
i
k
i
w
wxclassxgx
N
I
i
i
:
1
1
2
(2)
C
i
wiW
i
IN
N
I
1
1
(3)
C
i
iiB
gN
N
I
1
2
1
(4)
Where
2
. is the square of the Euclidean
norm:
t
xxx
. Global inertia
G
I (see definition
(1)) is computed over the entire data set. In contrast,
within-category inertia
W
I (see definitions (2) and
(3)) is the weighted sum of the inertia computed on
each category where
i
g
represents the center of
gravity of patterns belonging to category
i
w , where
the weighting is the a priori probability of each
category (
i
N is representing the number of patterns
belonging to category
i
w ). Between-category inertia
B
I (see definition (4)) is computed on the centers of
gravity of each category.
3.2 Dispersion and Fisher Criterion
Generally speaking, in a supervised classification
problem, classification performance depends on the
discrimination power of the features, that is to say,
the set of input dimensions which compose the
patterns of the data set. Dispersion and the Fisher
criterion are two measures (Blayo et al., 1995) for
the discrimination between classes (categories
defined in the input space). The overlapping rate
between categories is measured by the Fisher
criterion (see expression (5)). In addition, a simple
measure for the dispersion between categories is the
mean dispersion of category
i
w in category
j
w defined in expression (6). It is important to note
that, similar to conditional probabilities, the
dispersion matrix is not symmetric.
W
B
I
I
FC
(5)
Cji
I
gg
D
j
w
ji
ij
,1
(6)
As it can be deduced, the discrimination is better if
the Fisher criterion is large. Similarly, if the
dispersion measure (6) between two categories is
large then these categories are well separated and the
between category distance is larger than the mean
dispersion of the classes. Furthermore, if this
measure is close to or lower than one, the categories
are highly overlapped. In order to apply the Fisher
criterion and dispersion measures the data set is
normally pre-processed using a linear re-scaling
(Bishop, 1995) so as to arrange all the input
dimensions to have similar values. In addition, it is
important to note that a high degree of overlapping
between two categories does not necessarily imply
significant confusion between them from the
classification point of view. For instance, that is the
case for multimodal or very elongated categories
.
3.3 Confusion Matrix
The confusion matrix (Blayo et al., 1995) is a
structural parameter of a data set which provides an
estimation of the probability for patterns of one
category to be attributed to any other or to the
original category. Furthermore, it provides a generic
measure of classification complexity. Let us denote
as
the random variable describing the patterns of
StructuralAnalysisofNuclearMagneticResonanceSpectroscopyData
215

the data set. Supposing
is a discrete variable, the
confusion matrix can be defined as shown below,
where
f
is a discriminating function:
k
kikij
fwpC /
(7)
More specifically, a classifier is always defined in
terms of its discriminating function
f which
divides the d-dimensional input space into as many
regions as there are categories. If there are C
categories
i
w , Ci 1 , the discriminant function
may also be expressed in terms of the following
indicator function
i
f , where 1)(
uf
i
if
1)( uf
and 0)( uf
i
otherwise. The classifier
performance may also be expressed by the averaged
classification error:
C
i
C
j
iji
fCpfE
11
)(
(8)
The best confusion matrix is that corresponding to
the Bayesian classifier (minimal attainable
classification error). It can be deduced that the
confusion matrix cannot be computed using the
Bayesian classifier as it would imply a perfect
knowledge of the statistics of the problem
(conditional probabilities
i
wp /
and the a priori
probabilities
i
p ). Therefore, the best confusion
matrix must be in practice approximated. To this
end, the k-nearest neighbour classifier (Bishop,
1995); (Blayo et al., 1995); (Fukunaga, 1990) is
often used because of its powerful probability
density estimation properties. More specifically, a
set of values for k are generated, for instance the
following odd sequence k = 1,3,5,7,9,11.
Afterwards, a leave-one-out cross-validation
procedure (Haykin, 1999); (Fukunaga, 1990) is
performed for the entire dataset for each k from the
selected set of values. Finally, the best confusion
matrix is that obtained for the value of k which
minimizes the performance error defined in
expression (8).
4 EXPERIMENTS
In this section we consider that the FIDs (Free
Induction Decays) have already been Fourier
Transformed to the frequency domain, and the
artefact removal stage carried out. Furthermore, we
suppose the data is arranged into a matrix in which
each row corresponds to a spectral sample, where
two consecutive rows correspond to signal frames
taken with a time difference equal to the repetition
time of the NMR sequence and each column to a
metabolic signal. The metabolic signal corresponds
to the spectral intensity at a particular chemical shift.
In addition, it is important to note that the metabolic
signals (columns in the data matrix) have values that
differ significantly, even by several orders of
magnitude. Additionally, there are correlations
between them (sample dimensions) due to the
spectral overlap caused by the proton homonuclear
scalar coupling. To take into account the differences
in magnitude of metabolic signals, but allowing at
the same time the possibility to exploit the existing
correlations between them, the whitening transform
(Fukunaga, 1990); (Bishop, 1995) was used to
normalize all the data sets used in this section.
4.1 Data Set Description
For experimental purposes two data sets were used
both of them corresponding to short-TE NMR single
voxel brain spectroscopy. Let us denote the first data
set as A, which corresponds to data collected from
11 healthy patients of ages ranging from 25 up to 45,
with a mean of 31.45 years. The data was collected
from different brain regions (see table 1 for details)
and with approximately equal voxel sizes. The
acquisition time was approximately equal to 5
minutes for all patients, using a total echo time (TE)
equal to 23ms and a repetition time (TR) of 1070ms.
Similarly, the second data set (see table 2 for
details) is a small data set used for comparison
purposes in the experimental settings of section 4.2
and it is composed of data also collected at short TE
but with a slightly different parameterization. Let us
denote the second data set as B. For these data, the
total echo time was set to 35ms and the repetition
time to 1500ms and the patients´ ages ranged from
30 up to 45 with a mean of 36 years. In addition, the
data corresponds to three patients, where data matrix
B
1
and B
3
belong to two healthy patients, while data
matrix B
2
corresponds to a patient that was
diagnosed with a tumour (after a rigorous clinical
diagnosis procedure including biopsy). In particular,
the data matrix corresponding to patient B
2
represents data obtained exactly from the brain
tumour area.
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
216

Table 1: Data set A. Illustration of the most relevant
characteristics of the dynamic data set used for the first
experimental setting. This data set is composed of NMR
data collected using a total echo time (TE) equal to 23ms
and a repetition time (TR) of 1070ms from 11 healthy
patients. Each data matrix A
i
(i =1,2,..,11) is composed
approximately of 300 rows and 1068 columns. In other
words, the samples belong to an input space of 1068
dimensions. The notation used for the voxel location in
NMR Spectroscopy is “Left-Right” (L/R) for the “x”
dimension, “Anterior-Posterior” (A/P) for the “y”
dimension and “Inferior-Superior” (I/S) for the “z”
dimension. Voxel dimensions are expressed in
millimetres.
Data Set A
TE = 23ms
TR= 1070ms
Voxel Location Voxel Size
[A
1
] [L,P,I]=[0.9,6.3,17.2] [20,20,20]
[A
2
] [L,P,S]=[8.1,27.7,51.5] [29,20,27]
[A
3
] [L,P,S]=[6.7,9.5,15.1] [20,20,20]
[A
4
] [L,P,S]=[0.3,6.6,44.9] [20,20,20]
[A
5
] [L,P,S]=[1.2,18.5,61.6] [20,20,20]
[A
6
] [R,P,S]=[0.3,14.6,68.8] [20,20,20]
[A
7
] [L,P,S]=[2.79,25.97,60.9] [20,18,15]
[A
8
] [R,P,S]=[21.3,92.8,43.2] [20,20,20]
[A
9
] [L,P,S]=[27.4,25,63.8] [20,20,20]
[A
10
] [L,P,S]=[23.2,26.1,38.7] [20,20,20]
[A
11
] [R,P,S]=[4.1,13.3,45.1] [29,20,27]
Table 2: Data set B. Illustration of the most relevant
characteristics of the dynamic data set used for the second
experimental setting. This data set is composed of NMR
data collected from 3 patients using a total echo time (TE)
equal to 35ms and a repetition time (TR) of 1500ms. Each
data matrix B
i
(i =1,2,3) is composed approximately of
200 rows and 1068 columns. The data matrix B
2
corresponds to a patient who was diagnosed with a
tumour. The rest of data correspond to healthy patients.
The notation used for voxel location and the units used for
the voxel size are identical to those used in table 1.
Data Set B
TE = 35ms
TR= 1500ms
Voxel Location Voxel Size
[B
1
] [L,A,S]=[19.4,14.8,96.3] [16.5,17.7,17]
[B
2
] [R,A,S]=[20.9,35.5,76.2] [20,29.6,20]
[B
3
] [L,A,S]=[30,16,64.7] [20,20,20]
All the spectral data were generated and pre-
processed using a spectroscopic and processing
software package from GE Medical Systems
(SAGE). This tool comes with a set of built-in
functions (macro reconstruction operations) which
provide different useful processing options of raw
FID data. We used a macro reconstruction operation
which provides internal water referencing, spectral
apodization, zero filling, convolution filtering and
Fourier transform operation on each of the acquired
frames. However, it is important to note that the
convolution filtering and water suppression options
were not selected. The result of this processing step
is a data matrix where each column represents a
temporal series spectrum of a specific metabolic
signal and each row represents a sample or pattern
from the brain region of interest. Each sample
belongs to a space of 1068 dimensions
corresponding to a chemical shift range of [-0.8, 4.3]
ppm. As mentioned in section 2, this interval
corresponds to the range where the main resonances
concerning the 35 known metabolites involved in
brain metabolism are located.
4.2 Experimental Results
The first experiment conducted was designed to
check the variance of the measures corresponding to
different healthy subjects. At this point, it is
important to remember that both data sets described
in the previous section are dynamic. In addition, the
set of parametric measures introduced in section 3.2
were proposed in a supervised learning context. In
this kind of machine learning paradigm knowledge
about the problem is represented by means of input-
output examples, specifically, examples in the form
of vector of attribute values and known classes. This
means that the samples of the data set must be rated
as belonging to a predefined set of categories. In our
case, the categories are defined according to the
number of different subjects that compose the
database. For data set A there are samples coming
from eleven different individuals, therefore
according to the proposed schema we have eleven
different categories for the samples. Moreover,
samples belonging to subject A
i
are rated as
belonging to the class C
i
where i = 1,2,…11. At this
point, it is important to highlight the fact that we
have chosen this categorization scheme for two
reasons: firstly, as stated above, in order to check the
performance of the proposed structural parameters
and secondly, because of a lack of data from patients
presenting disorders that could bias the results.
Ideally, for diagnosis purposes we would have used
just two categories for discriminating disease.
Table 3 (see the appendix for details), shows the
results obtained after computing the dispersion for
data set A after the categorization procedure
described above. The first thing to note is that most
of the dispersion values are below one, thereby
indicating a high degree of overlapping between
classes. These results would suggest that the absence
of substantial differences from data collected from
different patients and brain regions is a plausible
indication of the existence of similar metabolic
processes. It is important to emphasize that
StructuralAnalysisofNuclearMagneticResonanceSpectroscopyData
217

metabolic processes associated to tumour cells are
radically different when compared to those of the
original non-transformed cell types.
However, it is important to note that the
dispersion values associated with categories C
2
and
C
11
with respect to the rest of the categories (rows
and columns A
2
and A
11
of the data matrix) are
slightly higher when compared with the rest of the
elements of the matrix. Indeed, there are dispersion
values which are close to unity or even slightly
higher than unity. A careful analysis revealed that
this effect was caused by the voxel size. The size of
the voxel for the “x” and “z“ dimensions (see table
1) is slightly bigger for categories C
2
and C
11
with
respect to the standard voxel size [20,20,20]. This is
also true for dimensions “y” and “z” on the voxel
associated with class C
7
. We observed that the effect
is to some extent proportional to the discrepancy
between the actual size and the standard voxel size.
In order to further validate the results obtained with
the dispersion matrix we computed the Fisher
criterion obtaining a value of 0.8504 which
confirmed the expected overlapping between
classes.
In addition, following a similar procedure we
computed the confusion matrix associated with the
eleven categories composing data set A. Table 4 (see
the appendix for details) shows the best confusion
matrix (see section 3.3) obtained by the KNN
classifier following a leave-one-out statistical cross-
validation procedure. It is important to note that the
estimations of conditional probabilities between
classes shown in the table are multiplied by a factor
of 100 to get percentage values. Therefore, it is easy
to deduce that there is no apparent confusion
between categories as most of the values are zero or
close to zero. Nevertheless, samples belonging to
class C
6
are apparently the most difficult to classify.
Generally speaking, a high degree of overlapping
given by the dispersion does not necessarily mean
significant confusion between the classes from a
classification point of view. This is an indication of
the existence of multi-modal or very elongated
categories.
The second experiment conducted was designed
to check the sensitivity of the structural parameters
introduced in section 3.2 for detecting the existence
of metabolic changes as a result of including data
samples of a patient who was diagnosed with a
tumour. In turn, this experimental setting also
permitted to assess the influence of the parameters
of the PRESS sequences used. To this end, we
merged the two available data sets A and B (see
section 4.1 for details) to create a unique database.
We followed the same categorization scheme
explained before consisting of assigning as many
categories as the number of patients, where data
samples belonging to the same patient were assigned
to the same category.
Let us denote the merged data set as A+B. It is
important to remember that the samples associated
with data set B were collected using a different
parameterization sequence from that used for data
set A. In particular, for data set B the echo time used
was 35ms and the repetition time 1500 ms. The
fisher criterion computed for this data set led to a
value of 0.8262 indicating overlapping between the
defined categories.
Table 5 (see the appendix for details) shows the
results of computing the dispersion matrix for the
data set A+B. The first thing that can be gleaned
from the table is that most of the values are below
one, thereby indicating the existence of overlapping
between classes from the dispersion point of view.
Therefore, from a dispersion point of view there is
not too much difference between data samples
coming from the two different parameterizations,
although this can be considered to some degree a
logical result taking into account that the echo time
of the two sequences are relatively close.
Nevertheless, the samples associated with the class
representing a disorder (i.e., a patient with a tumour)
led to dispersion values much higher than for the rest
of the values found in the table, even taking into
account the voxel size effect. More specifically, they
are very close or bigger than two (see column B
2
from table 5) indicating a dispersion caused by the
existence of the disease. At this point, it is important
to remember that the dispersion matrix is not
symmetric. From a geometrical point of view,
according to equations (6) and (10) the strong
dispersion presented by the data samples
representing a disorder (i.e., a tumour) with respect
to the rest of samples indicated a reduced distance in
average from these samples to the centroid of their
category (i.e., category B
2
). In addition, these results
seem to indicate that classes from healthy patients
are well separated from the class indicating a
disease. Although not shown here, this was also
confirmed by the confusion matrix. Specifically,
from a classification point of view there is no
apparent confusion between the category
representing the patient with a tumour and the other
categories.
Finally, despite our previous considerations
concerning the amount of data, in order to deepen
and further complement the previous results, we
conducted an experiment consisting of using the
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
218

entire database A+B but now defining only two
categories C
0
and C
1
to indicate “health” and
“disease” respectively. The categorization procedure
is similar to the procedure described above. All the
samples belonging to data matrix B
2
were rated as
belonging to class C
1
, while the rest of the samples
were rated as belonging to category C
0
. The results
of the dispersion matrix and confusion matrix
computation are shown in table 6. From the
inspection of the table we appreciate that there is a
slight confusion for the recognition of category C
1
(“disease”). Specifically, there is a certain
probability that patterns belonging to the class
“health” may be rated as belonging to the class
“disease”, although this probability is small, and
from a diagnosis point of view an error of this kind
would be less serious when compared to the
opposite case which is apparently inexistent.
To summarize, tumour cells exhibit radical
genetic, biochemical and histological differences
with respect to the original non-transformed cell
types. We have shown that the fact of using the
dynamical aspects of NMR data together with
relatively simple structural measure like dispersion
and Fisher criterion could substantially help the
diagnosis procedure of the clinical investigator.
Indeed, the aforementioned structural measures used
in a non-supervised learning context have proven to
be very sensitive not only to discrepancies on the
specific parameterizations of the NMR measuring
process (i.e., voxel size) but also to intrinsic
anomalies presented by the NMR data as a result of
metabolic changes in the brain tissue studied. At this
point, it is important to remember that current
clinical diagnosis procedures do not make use of the
dynamical aspects of NMR-based data. Indeed, they
use averaged data (i.e, mean value of the data matrix
of tables 1 and 2). Hence, these results would
invalidate the traditional view that disregards the
dynamic aspects of MRS data as being devoid of
information. This methodical reasoning had been
also previously suggested in (Chinea, 2011) where it
was shown the information-richness associated to
the temporal dynamics of NMR metabolic signals as
a result of its chaotic nature.
Moreover, if the tumour size is relatively small
when compared to the voxel size the fact of using
averages could mask the presence of a tumour.
Although further experiments must be carried out,
for instance using a much larger amount of data,
these preliminary results would suggest that the
proposed structural measures for characterization of
spectral data could be used for the development of
non-invasive early tumour detection techniques. In
addition, these results would also provide a starting
point for the application of more sophisticated
machine learning techniques (Kolen and Kremer,
2001); (Schölkopf et al., 1999) to dynamic NMR
spectroscopy data.
5 CONCLUSIONS
In this paper we have investigated the application of
machine learning techniques to characterize
magnetic resonance spectroscopy data. Throughout
this paper we have focused explicitly on the
characterization of dynamic brain MRS data. We
have presented a formal description of the problems
associated with MRS-based data in terms of its
application context in the field of clinical diagnosis
research. We have shown that the fact of using the
dynamical aspects of NMR data together with
relatively simple structural measures (e.g.,
dispersion, Fisher criterion etc) could substantially
help the diagnosis procedure of the clinical
investigator. Indeed, the aforementioned structural
measures have proven to be very sensitive not only
to discrepancies on the specific parameterizations of
the NMR measuring process (e.g., voxel size) but
also to intrinsic anomalies presented by the NMR
data as a result of metabolic changes in the brain
tissue studied.
Summarizing, traditional clinical diagnosis
methods are characterized by working with averaged
data; conversely, this work attempts to identify
properties of the underlying dynamics using simple
structural measures that were shown to be very
sensitive to metabolic changes as those exhibited by
tumour cells. The principal advantage of the
proposed methodology is not only its simplicity but
also its ability to cope with the high-dimensional
characteristics of spectroscopy patterns which
allowed us to extract the relevant information
required for the detection and diagnosis of disease.
Moreover, the framework presented here opens
the possibility not only of having a starting point for
the use of more complex machine learning
techniques but also for the development of reliable
non-invasive diagnosis techniques using relatively
small experimental data sizes. We hope to have
provided sufficient motivation for further studies
and applications as we believe it is a great challenge
to adopt these methods and to apply them in the
clinical research field.
StructuralAnalysisofNuclearMagneticResonanceSpectroscopyData
219

REFERENCES
Aydin K., Ucok A., Cakir S. (2007), Quantitative Proton
MR Spectroscopy Findings in the Corpus Callosum of
Patients with Schizophrenia Suggest Callosal
Disconnection, AJNR American Journal of Radiology,
28, 1968-1974.
Basso K., M A. A., Stolovitzky G., et al. (2005), Reverse
engineering of regulatory networks in human B cells,
Nature Genetics, 37, 382-390.
Bishop C. M. (1995). Neural Networks for Pattern
Recognition, Oxford University Press, Oxford.
Bishop C. M., (2006). Pattern Recognition and Machine
Learning, Springer Science+Business Media, LLC.
Blayo F., Cheneval Y., Guérin-Dugué A., et al. (1995),
Enhanced Learning for Evolutive Neural Architecture,
ESPRIT Basic Research Project Number 6891,
Deliverable R3-B4-P, Task B4 (Benchmarks), pp. 11-
22.
Bruschweiler R., Zhang F. (2004), Covariance nuclear
magnetic resonance spectroscopy, Journal of
Chemical Physics 120, 5253-5261.
Cherkassy V., Mulier F. M. (2007), Learning from Data:
Concepts, Theory and Methods 2nd Edition, Wiley,
New Jersey, pp.92-127.
Chinea A. (2011), Nonlinear Dynamical Analysis of
Magnetic Resonance Spectroscopy Data, Lecture
Notes in Computer Science 6636, 469-482.
Cloarec O., Dumas M. E., Craig A., et al. (2005),
Statistical Total Correlation Spectroscopy: An
Exploratory Approach for Latent Biomarker
Identification from Metabolic 1H NMR Data Sets,
Analytical Chemistry 77, 1282-1289.
Ebbels T. M. D., Cavill R., Bioinformatic methods in
NMR-based metabolic profiling (2009), Progress in
Nuclear Magnetic Resonance Spectroscopy 55, 361-
374.
Frahm J., Hanioke W., Merboldt K. D., Transverse
coherence in rapid FLASH NMR imaging , Journal of
Magnetic Resonance 72 (1987) 307-314.
Friedman N. (2004), Inferring Cellular Networks Using
Probabilistic Graphical Models, Science, 303, 799-
805.
Frye M. A., Watzl J., Banakar S., et al. (2007), Increased
Anterior Cingulate/Medial Prefrontal Cortical
Glutamate and Creatine in Bipolar Depression,
Neuropsychopharmacology 32, 2490-2499.
Fukunaga K. (1990), Statistical Pattern Recognition, 2nd
Edition, Academic Press, San Francisco.
Haykin S. (1999), Neural Networks: A Comprehensive
Foundation, Prentice Hall, New Jersey.
Jansen J. F. A., Backes W. H., Nicolay K., et al. (2006), H
MR Spectroscopy of the Brain: Absolute
Quantification of Metabolites, Radiology 240, 318-
332.
Keun H. C, Athersuch T. J., Beckonert O., et al. (2008),
Heteronuclear 19F−1H Statistical Total Correlation
Spectroscopy as a Tool in Drug Metabolism: Study of
Flucloxacillin Biotransformation, Analytical
Chemistry 80, 1073-1079.
Kolen J. F., Kremer S. C. (Eds.) (2001). A Field Guide to
Dynamical Recurrent Networks. IEEE Press,
Piscataway, New Jersey.
Kwock L., Smith J. K., Castillo M., et al. (2006), Clinical
role of proton magnetic resonance spectroscopy in
oncology: brain, breast, and prostate cancer, Lancet
Oncology 7, 859-868.
Lisboa P. J. G., Vellido A., Tagliaferri R., Napolitano F.,
et al. (2010), Data Mining in Cancer Research, IEEE
Computational Intelligence Magazine, vol. 5, 1 (2010)
14-18.
Nelson S. J., Brown T.R. (1987), A method for automatic
quantification of one-dimensional spectra with low
signal-to-noise ratio, Journal of Magnetic Resonance
75, 229-243.
Raamsdonk L. M., Teusink B., Broadhurst D., et al.
(2001), A functional genomics strategy that uses
metabolome data to reveal the phenotype of silent
mutations, Nature Biotechnology 19(1), 45-50.
Sadja P. (2006), Machine learning for detection and
diagnosis of disease, Annual Review of Biomedical
Engineering 8, 537-565.
Schölkopf B., Burges C. J. C., Smola A.J. (Eds.),
Advances in Kernel Methods—Support Vector
Learning, MIT Press, Cambridge, MA (1999), pp.
327–352.
Sigmundsson T., Maier M., Toone B. K. (2003), Frontal
lobe N-acetylaspartate correlates with
psychopathology in schizophrenia: a proton magnetic
resonance spectroscopy study, Schizophrenia
Research 64, 63-71.
Summerfield C., Gómez-Ansón B., Tolosa E., et al. (2002)
Dementia in Parkinson disease: a proton magnetic
resonance spectroscopy study, Archives of Neurology
59, 1415-1420.
Vanhamme L., Sundin T., Hecke P. V., et al. (2001), MR
spectroscopy quantitation: a review of time-domain
methods, NMR in Biomedicine 14, 233-246.
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
220

APPENDIX
Table 3: Illustration of the dispersion matrix computed for data set A when the categories (i.e., classes in a supervised
learning context) of the data samples are defined according to the number of different subjects that compose the database.
Specifically, samples belonging to subject Ai are rated as belonging to category C
i
. Dispersion values close to or lower than
one indicates a high degree of overlapping between the involved categories. Conversely, dispersion values bigger than one
provide an indication that these categories are well separated.
A
1
A
2
A
3
A
4
A
5
A
6
A
7
A
8
A
9
A
10
A
11
A
1
0 0.76 0.21 0.24 0.10 0.14 0.30 0.27 0.20 0.27 0.95
A
2
0.79 0 0.80 0.67 0.73 0.73 1.12 0.96 0.74 0.77 0.46
A
3
0.22 0.80 0 0.29 0.17 0.21 0.28 0.26 0.23 0.30 1.02
A
4
0.26 0.68 0.30 0 0.24 0.23 0.46 0.39 0.26 0.27 0.83
A
5
0.12 0.77 0.17 0.24 0 0.12 0.25 0.23 0.19 0.24 0.97
A
6
0.15 0.72 0.21 0.23 0.12 0 0.31 0.28 0.18 0.21 0.91
A
7
0.26 0.92 0.23 0.37 0.20 0.26 0 0.16 0.31 0.33 1.16
A
8
0.29 0.96 0.26 0.38 0.22 0.28 0.19 0 0.32 0.34 1.18
A
9
0.20 0.71 0.22 0.25 0.17 0.17 0.36 0.31 0 0.21 0.88
A
10
0.26 0.71 0.27 0.25 0.21 0.20 0.37 0.31 0.20 0 0.87
A
11
0.83 0.38 0.84 0.68 0.76 0.76 1.17 0.98 0.76 0.79 0
Table 4: Best confusion matrix obtained by the KNN classifier computed for the data set A with a Leave One Out statistical
cross-validation method when the categories of the data samples are defined according to the procedure described in table 3.
The values shown in the table corresponding to the estimation of conditional probabilities were multiplied by a factor of
100 to get percentage values. These results show that there is no apparent confusion between the defined categories.
A
1
A
2
A
3
A
4
A
5
A
6
A
7
A
8
A
9
A
10
A
11
A
1
100 0 0 0 0 0 0 0 0 0 0
A
2
0 99.45 0 0 0 0 0 0 0 0 0.54
A
3
0 0 100 0 0 0 0 0 0 0 0
A
4
0.52 0 0 98.94 0 0 0 0 0.52 0 0
A
5
8.42 0 0 0.52 91.05 0 0 0 0 0 0
A
6
14.73 0.52 0.52 1.05 1.57 79.47 0 0 1.05 1.05 0
A
7
0 0 0 0 0 0 100 0 0 0 0
A
8
0 0 0 0 0 0 1.57 98.42 0 0 0
A
9
0 0 0 0 0 0 0 0 100 0 0
A
10
0 0 0 0 0 0 0.52 0 0 99.47 0
A
11
0 0 0 0 0 0 0 0 0 0 100
Table 5: Dispersion matrix obtained when merging dynamic data sets A and B, both corresponding to short-TE NMR single
voxel brain spectroscopy. Most of the values are close to or lower to one indicating that categories involved are overlapped.
Nevertheless, the column associated to category B
2
presents a strong dispersion with respect to the rest of categories. The
sensitivity of the dispersion for this category is due to the fact that data samples corresponding to category B
2
correspond to
brain tissue that exhibited strong metabolic changes because of the tumour cells.
A
1
A
2
A
3
A
4
A
5
A
6
A
7
A
8
A
9
A
10
A
11
B
1
B
2
B
3
A
1
0 0.75 0.21 0.24 0.10 0.15 0.30 0.27 0.20 0.26 0.93 0.45 2.24 0.20
A
2
0.79 0 0.79 0.66 0.72 0.72 1.11 0.96 0.74 0.77 0.46 1.32 3.14 0.88
A
3
0.22 0.79 0 0.29 0.17 0.22 0.28 0.27 0.23 0.30 1.00 0.42 2.37 0.23
A
4
0.26 0.67 0.30 0 0.23 0.24 0.45 0.39 0.26 0.27 0.82 0.60 2.08 0.29
A
5
0.12 0.76 0.18 0.24 0 0.12 0.25 0.23 0.19 0.24 0.95 0.41 2.22 0.18
A
6
0.15 0.71 0.21 0.23 0.12 0 0.31 0.28 0.18 0.21 0.89 0.47 2.05 0.21
A
7
0.26 0.91 0.23 0.37 0.19 0.26 0 0.16 0.31 0.32 1.14 0.23 2.47 0.23
A
8
0.29 0.95 0.26 0.38 0.22 0.28 0.19 0 0.32 0.33 1.16 0.20 2.39 0.26
A
9
0.20 0.71 0.22 0.25 0.17 0.17 0.35 0.31 0 0.21 0.86 0.51 2.05 0.28
A
10
0.25 0.70 0.27 0.25 0.20 0.20 0.36 0.31 0.20 0 0.86 0.51 1.67 0.29
A
11
0.81 0.38 0.83 0.67 0.75 0.76 1.15 0.98 0.75 0.79 0 1.35 2.89 0.93
B
1
0.38 1.06 0.33 0.48 0.31 0.38 0.23 0.16 0.42 0.45 1.30 0 2.74 0.34
B
2
0.58 0.77 0.58 0.50 0.51 0.51 0.74 0.60 0.53 0.45 0.85 0.84 0 0.62
B
3
0.19 0.81 0.21 0.26 0.15 0.20 0.26 0.24 0.27 0.29 1.02 0.38 2.33 0
StructuralAnalysisofNuclearMagneticResonanceSpectroscopyData
221

Table 6: Dispersion and confusion matrixes after merging dynamic data sets A and B and considering only two categories
of samples in the input space: “health” and “disease” respectively. In particular, samples belonging to healthy patients are
rated as belonging to category C
0
("health") and the rest of samples (i.e., samples belonging to data matrix B
2
) are rated as
belonging to category C
1
("disease"). Once again the dispersion is sensitive to the resulting metabolic differences presented
by the brain tissue. From a classification point of view there is no apparent confusion between both categories.
Dispersion
Matrix
C
0
C
1
C
0
0 1.9655
C
1
0.1249 0
Confusion
Matrix
C
0
C
1
C
0
95.2721 0
C
1
4.7279 100
BIOINFORMATICS2013-InternationalConferenceonBioinformaticsModels,MethodsandAlgorithms
222
