
COMBINING GENE EXPRESSION AND CLINICAL DATA TO
INCREASE PERFORMANCE OF PROGNOSTIC BREAST CANCER
MODELS
Jana
ˇ
Silhav´a and Pavel Smrˇz
Faculty of Information Technology, Brno University of Technology, Boˇzetˇechova 2, 612 66 Brno, Czech Republic
Keywords:
Generalized linear models, Logistic regression, Regularization, Combined data, Gene expression data.
Abstract:
Microarray class prediction is an important application of gene expression data in biomedical research. Com-
bining gene expression data with other relevant data may add valuable information and can generate more
accurate prognostic predictions. In this paper, we combine gene expression data with clinical data. We use
logistic regression models that can be built through various regularized techniques. Generalized linear models
enables combining of these models with different structure of data. Our two suggested approaches are evalu-
ated with publicly available breast cancer data sets. Based on the results, our approaches have a positive effect
on prediction performances and are not computationally intensive.
1 INTRODUCTION
Microarray class prediction (Amaratunga and Cabr-
era, 2004) is an important application of gene expres-
sion data in biomedical research. Microarray experi-
ments monitor gene expressions associated with dif-
ferent phenotypes. Prediction of prognosis based on
different phenotypes is challenging due to relatively
small number of samples and high-dimensionality of
gene expression data. Combining gene expression
data with other relevant data may add valuable infor-
mation and can generate more accurate predictions.
In this paper, we combine gene expressions with
clinical data. Clinical data is heterogeneous and mea-
sures various entities (e.g. lymph nodes, tumor size),
while gene expression data is homogeneous and mea-
sures gene expressions. We assume that the combina-
tion of gene expressions with clinical data can involve
complementary information, which may yield more
accurate (disease outcome) predictions than those ob-
tained based on the use of gene expression or clinical
data alone. In literature, there are studies aimed at in-
tegrative prediction with gene expression and clinical
data, e.g. see (Li, 2006) and (Gevaert et al., 2007).
On the other side, redundant and correlated data can
have contradictory impact on prediction accuracy.
Methods combining biomedical data can be di-
vided into categories depending on the stage of inte-
gration (Azuaje, 2010). We propose an approach that
combines data at the stage of late integration, which
extends a part of work of (
ˇ
Silhav´a and Smrˇz, 2010).
We use logistic regression models that can be built
through various regularized techniques and can be ap-
plied to high-dimensional data as well. A key to com-
bining gene expression and clinical data is a frame-
work of generalized linear models (GLMs), which is
offered for many statistical models.
Simple logistic regression has been widely used
with clinical data in clinical trials to determinate the
relationship between variables and outcome and to
assess variable significance. Clinical data is usually
low-dimensional because gene expression data sets
include just a few clinical variables. That is why
we use simple logistic regression models with clin-
ical data and regularized logistic regression models
with high-dimensional gene expression data.
According to (Li, 2006), the penalized estimation
methods for integrative prediction and gene selection
are promising but computationally intensive. We ex-
perimented with R packages ‘mboost’, ‘glmnet’, ‘gr-
plasso’, ‘glmpath’ that regularize high-dimensional
data with penalties and at the same time these sta-
tistical models were developed for fitting in GLM
framework. R packages ‘mboost’ and ‘glmnet’ per-
formed very well and models fitting were not time-
consuming. We built the algorithms from these R
packages in our classifiers that combine gene expres-
sion and clinical data. In case of R package ‘mboost’,
we use a version of boosting that utilizes componen-
twise linear least squares (CWLLS) as a base proce-
589
Šilhavá J. and Smrž P..
COMBINING GENE EXPRESSION AND CLINICAL DATA TO INCREASE PERFORMANCE OF PROGNOSTIC BREAST CANCER MODELS.
DOI: 10.5220/0003881505890594
In Proceedings of the 4th International Conference on Agents and Artificial Intelligence (SSML-2012), pages 589-594
ISBN: 978-989-8425-95-9
Copyright
c
2012 SCITEPRESS (Science and Technology Publications, Lda.)

dure, that closely corresponds to fitting a logistic re-
gression model (B¨uhlmann and Hothorn, 2007). R
package ‘glmnet’ is an application of elastic net (Zou
and Hastie, 2005), which is a regularization and vari-
able selection method that can include both L
1
and L
2
penalties. The algorithms for fitting GLMs with elas-
tic net penalties were developed by (Friedman et al.,
2007), which also described logistic regression model
with elastic net penalties. Our approaches that com-
bine gene expression and clinical data improve pre-
diction performances and are not computationally in-
tensive.
The rest of this paper is organized as follows: The
relevant models with setting of their parameters and
the proposed approaches that combine data are de-
scribed in Section 2. Section 3 presents evaluation
methodology and results via comparative boxplots of
breast cancer data sets. It also includes a comparison
of execution times of applied approaches. This paper
is concluded in Section 4.
2 METHODS
Notation: Let X be p× n gene expression data matrix
with an element x
ij
, p genes and n samples. Let Z be
q× n clinical data matrix with an element z
ij
, q clini-
cal variables and n samples. y is n×1 response vector
with an element y
i
and with ground truth class labels
y ∈ {A, B}, where A and B can denote poor and good
prognosis. In the following text, the upper indexes
X, Z, distinguish from variables with gene expression
data, clinical data.
2.1 Generalized Linear Models
GLMs (McCullagh and Nelder, 1989) are a group of
statistical models that model the response as a non-
linear function of a linear combination of the predic-
tors. These models are linear in the parameters. The
nonlinear function (link) is the relation between the
response and the nonlinearly transformed linear com-
bination of the predictors. We employ GLMs in data
combining due to nice shared properties such as lin-
earity. GLMs are generalization of normal linear re-
gression models and are characterized by the follow-
ing features:
1. Linear regression model:
η
i
= β
0
+
q
∑
j=1
β
j
x
ij
+ ε
i
, (1)
where i = 1, . . . , n. β are regression coefficients and ε
is a random mean-zero error term.
2. The link function:
g(y
i
) = η
i
, (2)
where g is a link function, i = 1, . . . , n. η
i
is a linear
predictor. Respectively y
i
= g
−1
(η
i
), where g
−1
is an
inverse link function.
2.2 Logistic Regression Model
We use linear logistic regression model with clinical
data. The linear logistic regression model is an ex-
ample of GLM, where the response variable y
i
is con-
sidered as a binomial random variable p
i
and the link
function is logistic:
η = log
p
1− p
. (3)
Logistic regression model with clinical data can be
described with the following equation:
g(y
i
) = η
i
= β
Z
0
+
q
∑
l=1
β
Z
l
z
il
, (4)
where i = 1, . . . , n. g is the link function (3). y
i
or p
i
are outcome probabilities P(y
i
= A|z
i1
, . . . , z
iq
).
2.3 Boosting Model
A boosting with componentwise linear least squares
(CWLLS) as a base procedure is applied to gene ex-
pression data. A linear regression model (1) is consid-
ered again. A boosting algorithm is an iterative algo-
rithm that constructs a function
ˆ
F(x) by considering
the empirical risk n
−1
∑
n
i=1
L(y
i
, F(x
i
)). L(y
i
, F(x
i
)) is
a loss function that measures how close a fitted value
ˆ
F(x
i
) comes to the observation y
i
. In each iteration,
the negative gradient of the loss function is fitted by
the base learner. The gradient descent is an optimiza-
tion algorithm that finds a local minimum of the loss
function. The base learner is a simple fitting method
which yields as estimated function:
ˆ
f(·) =
ˆ
f(X, r)(·),
where
ˆ
f(·) is an estimate from a base procedure. The
response r is fitted against x
1
, . . . , x
n
.
The functional gradient descent (FGD) boosting
algorithm, which has been given by (Friedman, 2001)
is as follows (B¨uhlmann and Hothorn, 2007):
1. Initialize
ˆ
F
(0)
≡
n
∑
i=1
L(y
i
, a) ≡ ¯y. Set m = 0.
2. Increase m: m = m + 1. Compute the negative
gradient (also called pseudo response), which is
the current resudial vector:
r
i
= −
∂
∂F
L(y, F)|
F=
ˆ
F
(m−1)
(x
i
)
r
i
= y
i
−
ˆ
F
(m−1)
(x
i
), i = 1, . . . , n.
ICAART 2012 - International Conference on Agents and Artificial Intelligence
590

3. Fit the residual vector (r
1
, . . . , r
n
) to (x
1
, . . . , x
n
)
by base procedure (e.g. regression)
(x
i
, r
i
)
n
i=1
base procedure
−−−−−−−−−−−−→
ˆ
f
(m)
(·),
where
ˆ
f
(m)
(·) can be viewed as an approximation
of the negative gradient vector.
4. Update
ˆ
F
(m)
(·) =
ˆ
F
(m−1)
(·) + ν ·
ˆ
f
(m)
(·), where
0 < ν < 1 is a step-length (shrinkage) factor.
5. Iterate steps 2 to 4 until m = m
stop
for some stop-
ping iteration m
stop
.
The CWLLS base procedure estimates are defined as:
ˆ
f(X, r)(x) =
ˆ
β
ˆs
ˆx
ˆs
, ˆs = arg min
1≤ j≤p
n
∑
i=1
(r
i
− β
j
x
ij
)
2
,
ˆ
β
j
=
∑
n
i=1
r
i
x
ij
∑
n
i=1
(x
ij
)
2
, j = 1, . . . , p.
ˆ
β are coefficient estimates. ˆs denotes the index of the
selected (the best) predictor variable in iteration m.
For every iteration m, a linear model fit is obtained.
BinomialBoosting (B¨uhlmann and Hothorn,
2007), which is the version of boosting that we
utilize, use the negative log-likelihood loss function:
L(y, F) = log
2
(1 + e
−2yF
). It can be shown that this
population minimizer has the form (B¨uhlmann and
Hothorn, 2007): F(x
i
) =
1
2
log
p
1−p
, where p is
P(y
i
= A|x
i1
, . . . , x
ip
) and relates to logit function,
which is analogous to logistic regression.
2.4 Elastic Net Model
We also use elastic net (Zou and Hastie, 2005) with
gene expression data. The linear regression model (1)
is considered again. The elastic net optimizes the fol-
lowing equation with respect to β (Friedman et al.,
2010):
ˆ
β(λ) = arg min
1
2n
n
∑
i=1
(y
i
−
p
∑
j=1
β
j
x
ij
)
2
+ λ
p
∑
j=1
P
α
(β),
where: P
α
(β) = (1− α)
1
2
kβk
2
L
2
+ αkβk
L
1
or P
α
(β) =
(1− α)
1
2
β
2
j
+ α|β
j
| is the elastic net penalty.
P
α
(β) =
L
1
penalty if α = 1,
L
2
penalty if α = 0,
elastic net penalty if 0 < α < 1.
(5)
In our case, elastic net builds logistic regression
model with elastic net penalties. The regular-
ized equation is fitted by maximum (binomial)
log-likelihood and solved by coordinate descent,
see (Friedman et al., 2010). The coordinate update
has the form:
ˆ
β
j
←
S(
1
n
∑
n
i=1
x
i
r
ij
, λα)
1+ λ(1− λ)
=
S(β
∗
j
, λα)
1+ λ(1− λ)
, (6)
where r
ij
is the partial residual y
i
− ˆy
ij
for fitting
ˆ
β
j
and S(κ, γ) is the soft-thresholding operator, which
takes care of the lasso contribution to the penalty.
More detailed description is given in (Friedman et al.,
2007).
A simple description of CCD algorithm for elastic
net is as follows (Friedman et al., 2010):
The authors assume that the x
ij
are standardized:
∑
n
i=1
x
ij
= 0,
1
n
∑
n
i=1
x
2
ij
.
− Initialize all the
ˆ
β
j
= 0.
− Cycle around till convergence and coefficitents
stabilize:
1. Compute the partial residuals:
r
ij
= y
i
− ˆy
ij
= y
i
−
∑
k6= j
x
ik
β
k
.
2. Compute the simple least squares coefficient of
these residuals on jth predictor:
β
∗
j
=
1
n
∑
n
i=1
x
ij
r
ij
.
3. Update
ˆ
β
j
by soft-thresholding:
ˆ
β
j
← S(β
∗
j
, λ), which equals (6).
2.5 Combining Gene Expression and
Clinical Data
In GLMs, the linear models are related to the response
variable via a link function (2). For binary data, we
expect that the responses y
i
come from binomial dis-
tribution. Therefore, logit link function is used in all
models with clinical and gene expression data. η
i
is a
linear model, which is a linear part of logistic regres-
sion and a linear regression model in boosting with
CWLLS described in Subsection 2.3. We combine
the data by summing the linear predictions of clinical
and gene expression data:
η
i
= η
Z
i
+ η
X
i
. (7)
According to the additivity rule that is valid for linear
models, it is possible to sum the linear models:
η
i
= β
Z
0
+
q
∑
l=1
β
Z
l
z
il
+
p
∑
j=1
β
X
j
x
ij
. (8)
Then the inverse link function g
−1
, which is the in-
verse logit function, is applied to the sum of linear
predictions η
i
:
g
−1
(η
i
) = logit
−1
(η
i
) =
e
η
i
1+ e
η
i
. (9)
COMBINING GENE EXPRESSION AND CLINICAL DATA TO INCREASE PERFORMANCE OF PROGNOSTIC
BREAST CANCER MODELS
591
0.12 0.08 0.04 0.00
0.0 0.4 0.8
Lambda
AUC
(a) p = 20
0.12 0.08 0.04 0.00
0.0 0.4 0.8
Lambda
AUC
(b) p = 400
0.12 0.08 0.04 0.00
0.0 0.4 0.8
Lambda
AUC
(c) p = 1000
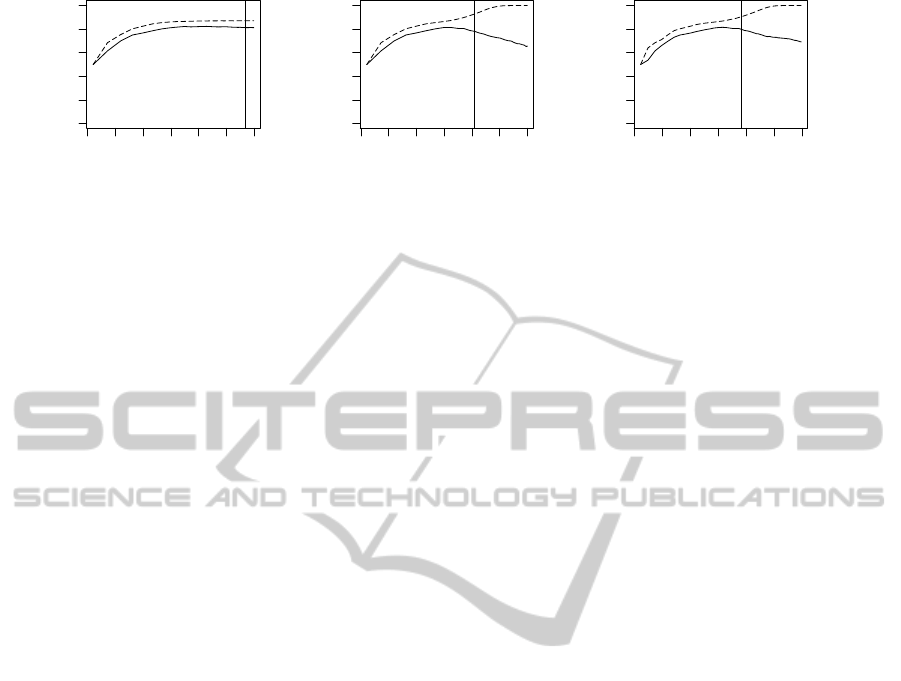
Figure 1: Examples of λ solution path produced by glmnet algorithm in the setting with the lasso penalty. The dashed (solid)
line denotes AUC estimated from training (test) data set. The vertical line denotes λ
OPT
.
For better readibility in results, we denote this ap-
proach LOG+B.
Similarly we combine logistic regression and reg-
ularized logistic regression models from elastic net.
For better readability in results, we denote this ap-
proach LOG+EN.
3 RESULTS
We tested the described approaches with simulated
and publicly available breast cancer data sets. How-
ever, the results with simulated data are not shown
due to the conference page limit. The performances
of individual models were evaluated as well because
of a comparison of the models.
In R environment, we used glm function from
‘base’ package to fit the logistic regression models
with clinical data; ‘mboost’ and ‘glmnet’ packages to
fit the logistic regression models with gene expression
data.
The shrinkage factor ν and the number of it-
erations of the base procedure are the main tun-
ing parameters of boosting. Based on recommen-
dation from (B¨uhlmann and Hothorn, 2007), we set
ν = 0.1 to the standard default value in ‘mboost’
package. The numbers of iterations were esti-
mated with Akaike’s information stopping criterion
(AIC) (Akaike, 1974). We also tested a functional-
ity of AIC stopping criterion, and evaluated perfor-
mances of data with fixed number of iterations within
the range 50-800 iterations, and compared with AIC
estimated performances (data not shown). The max-
imal number of iterations was set to m
max
= 700 and
was sufficient.
The choice of the penalty (5) is the main tuning
parameter of elastic net. The algorithm from ‘glm-
net’ package computes a group of solutions (regular-
ization path) for a decreasing sequence of values for
λ (Friedman et al., 2010). We evaluated all solutions
on training and test data set with different penalties
α (1, 0, 0.5) and with different numbers of variables
(20, 400, 1000) to inspect performances in different
dimensional setting. Based on results, we chose the
lasso penalty (α = 1) for further experiments. Fig-
ure 1 depicts examples of this experiment with the
lasso penalty. The vertical lines in the subfigures de-
note the estimated values of λ
OPT
, which were es-
timated via training data set cross-validation (CV).
The subfigures were generated from simulated gene
expression data set of moderate power, see (
ˇ
Silhav´a
and Smrˇz, 2010), and depict one Monte Carlo cross-
validation(MCCV) iteration (the same for all figures).
MCCV was applied as a validation strategy.
MCCV generates learning set in that way that the
learning data sets are drawn out of {1, ··· , n} samples
randomly and without replacement. The test data set
consists of the remaining samples. The random split-
ting in non-overlapping learning and test data set was
repeated 100-times. The splitting ratio of training and
test data set was set to 4 : 1. Responses consisted of
predicted class probabilities were measured with the
area under the ROC curve (AUC).
We test the described approaches in different set-
tings to simulate various quality of data sets. We con-
sidered redundant and non-redundant settings of data
and different predictive powers of gene expression
and clinical data. From the simulations, the combined
models make more accurate predictions or take over
the values of the models with higher performances.
We also evaluted the described approaches with
two publicly available breast cancer data sets. The
van’t Veer data set (van’t Veer et al., 2002) includes
breast cancer patients after curative resection. cDNA
Agilent microarray technology was used to give the
expression levels of 22483 genes for 78 breast cancer
patients. 44 patients that are classified into the good
prognosis group, did not suffer from a recurrencedur-
ing the first 5 years after resection, the remaining
34 patients belong to the the poor prognosis group.
The data set was prepared as described in (van’t Veer
et al., 2002) and is included in R package ‘DEN-
ICAART 2012 - International Conference on Agents and Artificial Intelligence
592
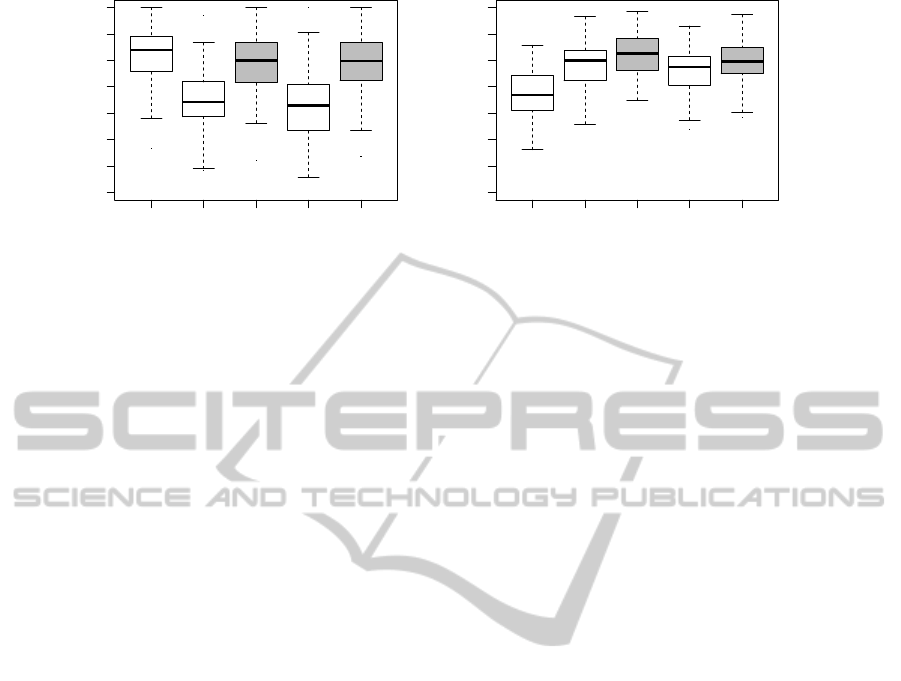
LOG B LOG+B EN LOG+EN
0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
Methods
AUC
(a) van’t Veer.
LOG B LOG+B EN LOG+EN
0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
Methods
AUC
(b) Pittman.
Figure 2: Breast cancer data sets. Boxplots of AUCs evaluated over 100 MCCV iterations.
MARKLAB’. The resulting data set includes 4348
genes. Clinical variables are age, tumor grade, es-
trogen receptor status, progesterone receptor status,
tumor size and angioinvasion. The second data set,
which is the Pittman data set (Pittman et al., 2004),
gives the expression levels of 12625 genes for 158
breast cancer patients. According to recurrence of
disease, 63 of these patients are classified into the
poor prognosis group, the remaining 95 patients be-
long to the good prognosis group. Gene expression
data was prepared with Affymetrix Human U95Av2
GeneChips. The data was pre-processed using pack-
ages ‘affy’ and ‘genefilter’ to normalize and filter the
data. The genes that showed a low variability across
all samples were clearedout. Theresulting data set in-
cludes 8961 genes. Clinical variables are age, lymph
node status, estrogen receptor status, family history,
tumor grade and tumor size.
Figure 2 depicts AUC box plots with the breast
cancer data sets. Considering the results with the
Pittman data set, the combined models have a pos-
itive effect on prediction performances and increase
AUCs. The combined models, built with the data of
van’t Veer, do not improve AUC performances and
it is better to use for prediction of prognosis clinical
data alone. The conclusion with the van’t Veer data
set also corresponds with findings, e.g. (Gruvberger
et al., 2003).
The performances of the combined models are
similar. LOG+B seems to perform slightly better than
LOG+EN with breast cancer data sets.
Execution Times
The execution times of the combined models are
mostly based on the execution times of the models
built with high-dimensional data, therefore we com-
pared the execution times of the FGD boosting algo-
rithm from the package ‘mboost’ (B,LOG+B) with
the CCD algorithm from the package ‘glmnet’ (EN,
LOG+EN). Figure 3 depicts the comparison. Increas-
ing numbers of variables are on the horizontal axes,
while total execution times for 100 MCCV iterations
(in minutes) are on the vertical axes. The plots indi-
cate that both methods achieve similar time values.
The execution times grow linearly. Besides, FGD
boosting grows with the number of boosting iterations
(in our simulations m
max=700
). A grid of 100 λ values
is computed in each iteration of EN. The simulations
were achieved with a standard PC (Intel T72500 Core
2 Duo 2.00 GHz, 2 GB RAM) and 32-bit operating
system.
4 CONCLUSIONS
In this paper, we combine gene expression and clini-
cal data to predict disease prognosis. We used logis-
tic regression models built by different ways. GLMs
enabled combining of these models. Two suggested
approaches were evaluated with simulated (data not
shown) and publicly available breast cancer data sets.
Both approaches performed well and showed similar
performances.
The accuracy of LOG+B has already been com-
pared with other methods from literature in (
ˇ
Silhav´a
and Smrˇz, 2010). It performed the same or better
than other methods from literature. LOG+EN can
be assessed analogously because of similar prediction
performance as LOG+B. The execution times of the
combined models grow linearly and the approaches
are not time consuming.
ACKNOWLEDGEMENTS
This work was supported by the Technology Agency
COMBINING GENE EXPRESSION AND CLINICAL DATA TO INCREASE PERFORMANCE OF PROGNOSTIC
BREAST CANCER MODELS
593
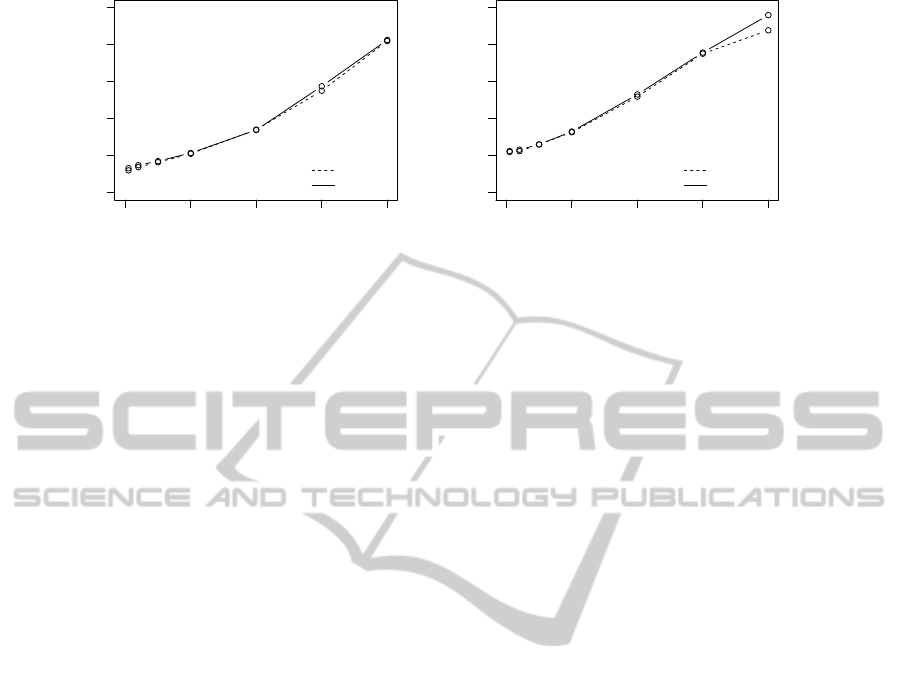
0 1000 2000 3000 4000
0 1 2 3 4 5
Number of genes [−]
Time [minutes]
B
LOG+B
(a) B and LOG+B.
0 1000 2000 3000 4000
0 1 2 3 4 5
Number of genes [−]
Time [minutes]
EN
LOG+EN
(b) EN and LOG+EN.
Figure 3: The comparison of computation times and their dependence on increasing number of variables. The graphs are
drawn for 100 MCCV iterations.
of the Czech Republic, project TA01010931 – GenEx
– System for support of the FISH method evaluation,
and the operational programme ’Research and De-
velopment for Innovations’ in the framework of the
IT4Innovations Centre of Excellence project, reg. no.
CZ.1.05/1.1.00/02.0070.
REFERENCES
Akaike, H. (1974). A New Look at the Statistical Model
Identification. IEEE Trans. Automat. Contr., 19 (6),
716-723.
Amaratunga, D. and Cabrera, J. (2004). Exploration and
Analysis of DNA Microarray and Protein Array Data.
John Wiley & Sons, Hoboken.
Azuaje, F. (2010). Bioinformatics and Biomarker Dis-
covery: “Omic” Data Analysis for Personalized
Medicine. John Wiley & Sons, Singapore.
B¨uhlmann, P. and Hothorn, T. (2007). Boosting Algorithms:
Regularization, Prediction and Model Fitting. Statist.
Sci., 22, 477-505.
Friedman, J., Hastie, T., H¨ofling, H., and Tibshirani, R.
(2007). Pathways Coordinate Optimization. Ann.
Appl. Stat., 1, 302-332.
Friedman, J. H. (2001). Greedy Function Approximation: A
Gradient Boosting Machine. Ann. Statist., 29, 1189-
1232.
Friedman, J. H., Hastie, T., and Tibshirani, R. (2010). Reg-
ularization Paths for Generalized Linear Models via
Coordinate Descent. Journal of Statistical Software,
33 (1), 1-24.
Gevaert, O., Smet, F. D., Timmerman, D., Moreau, Y.,
and Moor, B. D. (2007). Predicting the Prognosis of
Breast Cancer by Integrating Clinical and Microar-
ray Data with Bayesian Networks. Bioinformatics, 22
(14), 147-157.
Gruvberger, S. K., Ringner, M., and Eden, P. (2003). Ex-
pression Profiling to Predict Outcome in Breast Can-
cer: the Influence of Sample Selection. Breast Cancer
Res., 5(1), 23-26.
Li, L. (2006). Survival Prediction of Diffuse Large-B-Cell
Lymphoma Based on both Clinical and Gene Expres-
sion Information. Bioinformatics, 22(04), 466-471.
McCullagh, P. and Nelder, J. A. (1989). Generalized Linear
Models. Chapman and Hall.
Pittman, J., Huang, E., and Dressman, H. (2004). Integrated
Modeling of Clinical and Gene Expression Informa-
tion for Personalized Prediction of Disease Outcomes.
Proc.Natl.Acad.Sci., 101(22), 8431-8436.
ˇ
Silhav´a, J. and Smrˇz, P. (2010). Improved Disease Outcome
Prediction Based on Microarray and Clinical Data
Combination and Pre-validation. Biomedical Engi-
neering Systems and Technologies, 36-41.
van’t Veer, L. J., Dai, H., and van de Vijver, M. J. (2002).
Gene Epression Profiling Predicts Clinical Outcome
of Breast Cancer. Nature, 530-536.
Zou, H. and Hastie, T. (2005). Regularization and Variable
Selection via the Elastic Net. Journal of the Royal
Statistical Society, Series B, 67, 301-320.
ICAART 2012 - International Conference on Agents and Artificial Intelligence
594
