
AN EXTREME LEARNING MACHINE CLASSIFIER
FOR PREDICTION OF RELATIVE SOLVENT ACCESSIBILITY
IN PROTEINS
Saras Saraswathi, Andrzej Kloczkowski and Robert L. Jernigan
Department of Biochemistry, Biophysics and Molecular Biology
L. H. Baker Center for Bioinformatics and Biological Statistics, Iowa State University
112 Office and Laboratory Building, Ames, IA, 50011, U.S.A.
Keywords: Relative solvent accessibility, Support vector machine, Neural network, Extreme learning machine,
Prediction.
Abstract: A neural network based method called Sparse-Extreme Learning Machine (S-ELM) is used for prediction of
Relative Solvent Accessibility (RSA) in proteins. We have shown that multiple-fold gains in speed of
processing by S-ELM compared to using SVM for classification, while accuracy efficiencies are
comparable to literature. The study indicates that using S-ELM would give a distinct advantage in terms of
processing speed and performance for RSA prediction.
1 INTRODUCTION
Proteins perform a variety of important biological
functions that are imperative to the wellbeing of all
living things. Various factors determine protein
functions, such as, its native structure, the
information coded in its constituent amino acid
sequences, its reactions to the surrounding solvent
environment and the Relative Solvent Accessibility
(RSA) values of its residues and. Evaluating RSA
values will help to gain an insight into the structure
and function of a protein.
Protein structures and other related values such
as RSA can be experimentally determined by using
NMR spectroscopy or X-Ray crystallography. But
these methods can be expensive in terms of cost,
time and other factors. There is an urgent need to
process large amounts of data (spawned by advances
in biotechnology) accurately and speedily in order to
decipher the information buried in biological data,
since it is impractical to do it manually.
Computational methods such as machine learning
algorithms provide an alternate way by which we
can study this data in a cost and time efficient
manner. Still, accuracies and processing efficiencies
in existing methods are inadequate and there is a
need for improvement. This study endeavours to
attain a large gain in processing efficiencies.
RSA prediction has contributed to the study of
protein functions in many applications; to determine
protein hydration properties (Ooi, Oobatake,
Namethy, & Scheraga, 1987), identify temperature
sensitive residues that can be targeted for
mutagenesis and to study contact residue
information (Shen and Vihinen 2003), improve
secondary structure prediction (Adamczak, Porollo
& Meller, 2005) and for fold recognition and protein
domain (DOMpro) prediction (Cheng and Baldi,
2006). RSA values can be used to gauge degree of
solvent exposure of segments of globular proteins
(Carugo, 2003), to find residues with potential
structural or functional (ConSeq) importance
(Berezin, Glaser, Rosenberg, Paz, Pupko, Fariselli,
Casadio, & Ben-Tal, 2004), help with rationale
design of antibodies and other proteins to improve
binding affinities (David, Asprer, Ibana, Concepcion
& Padlan, 2007). In general RSA values can help to
achieve cost and time efficiencies in drug discovery
processes and help to gain a better understanding of
biological processes.
Probability profiles are used by Gianese, Bossa
& Pascarella (2003) to predict RSA values from
single sequence and Multiple Sequence Alignment
(MSA) data. Singh, Gromiha, Sarai & Ahmad
(2006) estimate RSA values from an atomic
perspective. Pollastri, Martin, Mooney & Vullo
364
Saraswathi S., Jernigan R. and Kloczkowski A..
AN EXTREME LEARNING MACHINE CLASSIFIER FOR PREDICTION OF RELATIVE SOLVENT ACCESSIBILITY IN PROTEINS .
DOI: 10.5220/0003086803640369
In Proceedings of the International Conference on Fuzzy Computation and 2nd International Conference on Neural Computation (ICNC-2010), pages
364-369
ISBN: 978-989-8425-32-4
Copyright
c
2010 SCITEPRESS (Science and Technology Publications, Lda.)

(2007) use homologous structural information to
improve RSA prediction. In addition, tertiary
structure predictions are increasingly being
augmented and improved with information derived
from secondary structure and RSA predictions.
Zarei, Arab & Sadeghi (2007) find that pairs of
residues can influence RSA prediction accuracy.
Knowledge-based tools which use machine
learning techniques and statistical theory can be
valuable in predicting RSA, especially in the
absence of evolutionary information or where
sequences are not well preserved. A number of
computational methods have been used for RSA
prediction, such as Neural Networks (NN) (Shandar
and Gromiha, 2002; Adamczak et al., 2005; Cheng,
Sweredoski, & Baldi, 2006; Huang, Zhu & Siew,
2006;). Pollastri, Baldi, Fariselli & Casadio (2002)
use RSA values of residues for scoring remote
homology searches and modelling protein folding
and structure using a bidirectional recurrent neural
network (ACCpro). Other methods include
Information Theory (Manesh, Sadeghi, Arab &
Movahedi, 2001), Multiple Linear Regression
Methods (Pollastri et al. 2002; Wagner et al. 2005),
Support Vector Machines (SVM) (Nguyen and
Rajapakse 2005) and fuzzy k-nearest neighbour
algorithm (Sim, Kim & Lee, 2005). Kim and Park
(2004) have used the SVMpsi and long range
interactions to improve RSA accuracy. Chen, Zhou,
Hu & Yoo (2004) compare five different methods,
decision tree (DT), Support Vector Machine (SVM),
Bayesian Statistics (BS), Neural Network (NN) and
Multiple Linear Regression (MLR) on the same data
set in order to compare the capabilities of different
methods in predicting RSA. They conclude that NN
and SVM are among the best methods for RSA
prediction.
More recently, Bondugula and Xu (2008)
combine sequence and structural information to
estimate RSA values (MUPRED) in order to predict
RSA. Petersen, Petersen, Andersen, Nielsen and
Lundegaard (2009) argue for the need of a reliability
score (Z-score) for measuring the degree of trust that
can be related to individual predictions. Meshkin
and Ghafuri (2010) use a two-step approach, using
feature selection on physico-chemical properties of
residues and Support Vector Regression (SVR) to
predict RSA.
We propose to use a new fairly new method
called Sparse Extreme Learning Machine (S-ELM),
based on neural networks, which is capable of
extreme speeds compared to traditional neural
networks while maintaining current classification
accuracies.
This paper is organized as follows. Section 2
briefly discusses the S-ELM algorithm and
characteristics of the RSA data. Section 3 discusses
the results of this study with performance
comparisons with SVM and NETASA methods
followed by conclusions in Section 4.
2 METHODS AND DATA
2.1 Extreme Learning Machine
Single Layer Feed-forward Network (SLFN), with a
hidden layer and an activation function possess an
inherent structure suitable for mapping complex
characteristics, learning and optimization. They have
applications in bioinformatics for solving various
problems like pattern classification and recognition,
structure prediction and data mining. The free
parameters of the network are learned from given
training samples using gradient descent algorithms
that are relatively slow and have many issues in
error convergence. A modified SLFN model called
an Extreme Learning Machine (ELM) has emerged
recently (Huang, Zhu, & Siew 2006), where it has
been proved theoretically that ELM can provide
good generalization performance and overcome
some of the problems associated with traditional
NNs such as stopping criterion, learning rate,
number of epochs and local minima. ELM has good
generalization capabilities and capacity to learn
extremely fast. The input weights are chosen
randomly but the output weights are calculated
analytically using a pseudo-inverse. Many activation
functions such as sigmoidal, sine, Gaussian or hard-
limiting functions can be used at the hidden layer
and the class is determined as the class which has
the maximum output value. A comprehensive
description of the S-ELM algorithm is given by
Huang et. al., (2006).
Even though the ELM algorithm requires less
training time, the random selection of input weights
affects the generalization performance when the data
is sparse or data is imbalanced. Suresh, Saraswathi
and Sundararajan (2010) and Saraswathi et al.
(2010) offer an improved version of ELM called the
Sparse-ELM (S-ELM) which gives better
generalization for sparse data. Hence, we use S-
ELM algorithm for predicting the RSA of proteins
where the imbalance in data varies with the different
threshold values used. S-ELM is also well suited for
RSA predictions of sequences whose structures have
not yet been determined and where there are no
homologs in existing sequences. The data is discus-
AN EXTREME LEARNING MACHINE CLASSIFIER FOR PREDICTION OF RELATIVE SOLVENT
ACCESSIBILITY IN PROTEINS
365
sed in detail in section 3.
We call the ELM algorithm for each of the
training data sets over several thresholds. We find
the optimal number of hidden neurons using a
unipolar sigmoidal activation function (lambda =
0.001) and perform K-fold (k = 5) validations. In K-
fold validation, the training set is separated into K-
groups. K-1 groups are used for training in each of
the K iterations and the model is tested on the
remaining K
th
group. The optimal parameters are
stored and used during the testing phase. The
performance of the S-ELM classifier and the time
taken to develop the RSA S-ELM classifier model is
compared with SVM using LIBSVM (Fan, Chen and
Lin, 2005) approach to show that the S-ELM
approach can achieve a slightly better performance
within a much shorter time. Five-fold cross
validation accuracies, processing time gains and
comparative studies are discussed in the results
section.
2.2 Data
Proteins consist of sequences of amino acid residues
that play a key role in determining the secondary and
tertiary structure of a protein. The sequential
relationship among the solvent accessibilities of
neighbouring residues can be used to improve the
results (although solvent accessibility is considered
evolutionarily less preserved than secondary
structure). We use binary values and a window size
of 8 to represent the amino acid sequences.
RSA of an amino acid residue is defined
(Mucchielli-Giorgi et al. 1999) as the ratio of the
solvent-accessible surface area of the residue
observed in the 3-D structure to that observed in an
extended tripeptide (Gly-X-Gly or Ala-X-Ala)
conformation. RSA is a simple measure of the
degree to which each residue in an amino acid
sequence is exposed to its solvent environment. For
our study, we consider the well-known Manesh data
set (Manesh, Sadeghi, Arab, & Movahedi, 2001)
which has a high imbalance with respect to the
number of samples per class (Table 1), where the
number of samples belonging to one class is much
lesser than the samples belonging to the other
classes.
The Manesh data set consists of 215 proteins, of
which 30 proteins (7545 residues) with variable
number of amino acid residues are used for classifier
model development and the remaining 185 proteins
(43137 residues) were used for evaluating the
generalization performance of the S-ELM classifier
through a 5-fold cross-validation model. The data in
the training and testing set are cast into two-class
and three-class problems (Table 1) by determining
whether the RSA value is below, between or above a
particular threshold. We use various % thresholds (0,
5, 10, 25, 50 for two-class and between 10_20 or
25_50 for three class), in order to compare our
results with those existing in literature. A residue is
considered as buried if its value is less than or equal
to the lower range, partially buried if it is between
the lower and the higher range and considered
exposed if its RSA value is higher than the range of
values (> 20 or > 50). The accuracy of the
predictions depend on the value of the thresholds
chosen and can vary widely with different residue
compositions in different proteins as discussed in the
results section.
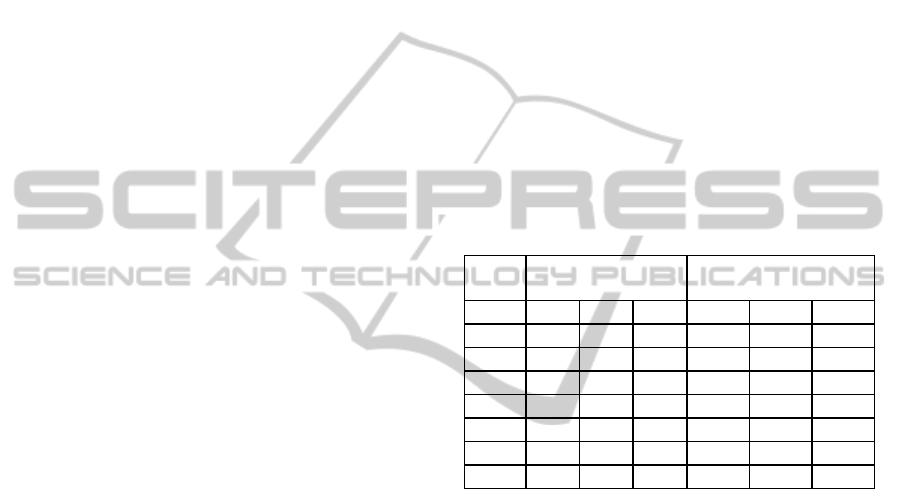
Table 1: Samples per class for 2-class and 3-class data
where thresholds are set between 0 and 50% for two class
(C0 and C1) and between 0, 10 and 50 % for 3-class (C0
C1 and C2).
Number of
Training residues
Number of
Testing residues
% C0 C1 C2 C0 C1 C2
0 867 6678 ** 4713 38424 **
5 5796 1749 ** 32943 10194 **
10 2826 4719 ** 15864 27273 **
20 4065 3480 ** 23111 20026 **
50 5796 1749 ** 32945 10192 **
10_20 3888 831 2826 22265 5008 15864
25_50 1750 1750 4065 10194 9832 23111
3 RESULTS AND DISCUSSION
We compare the results of our simulation using S-
ELM on the Manesh data set with the SVM
algorithm and NETASA (Shandar & Gromiha 2002)
methods (Figure 1 and Table 2), using the same set
of proteins for training and testing. Hence
comparisons with literature are made only with the
NETASA results.
The accuracy of the RSA predictions is measured
by the number of residues correctly classified as
belonging to class1 (E for exposed) for the two class
problem and as belonging to class2 (E) for the three
class problem. Prediction accuracy for training and
testing data sets is defined as the total number of
correctly predicted values for each class over the
total number of available residues in all classes. The
data shown in Table 2 indicates that the S-ELM
approach achieves a better accuracy for training and
testing than the corresponding results for the
ICFC 2010 - International Conference on Fuzzy Computation
366
NETASA paper. The SVM algorithm takes a longer
time to build the model as shown in Figure 2 and 3,
whereas the S-ELM algorithm process data at the
same speed for all combinations of data, showing
that the algorithm does not slow down when
complex data is involved. S-ELM uses optimal
parameters that are stored during the training phase
making it possible to run through the tests quickly.
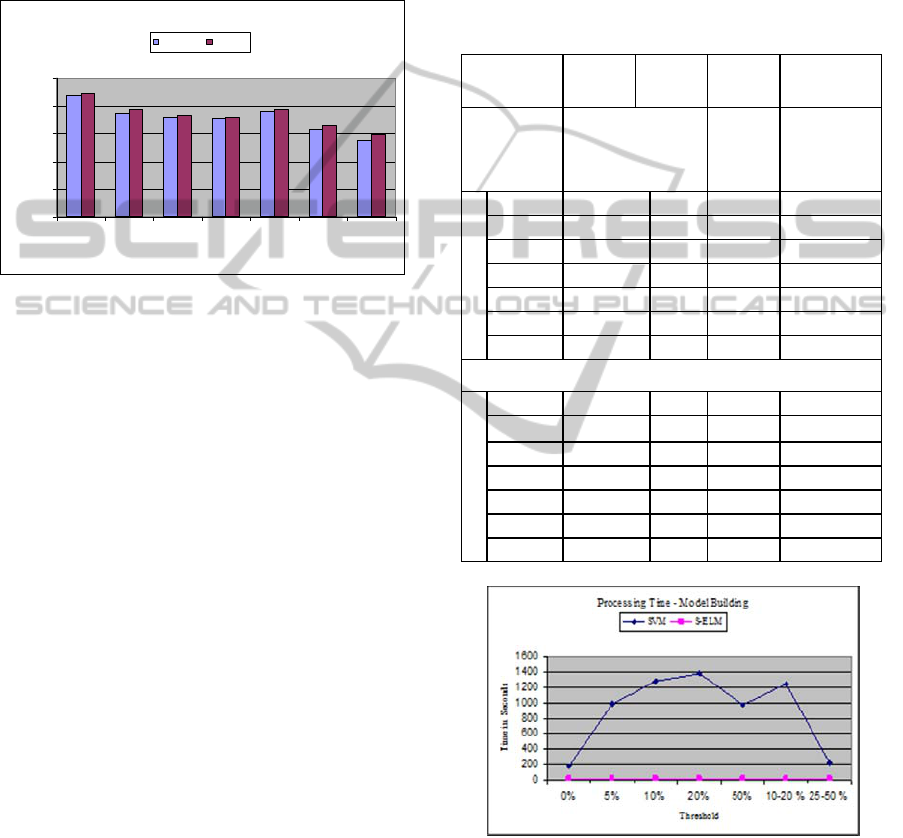
Figure 1: Accuracy comparison between NETASA and S-
ELM, shows slight improvements for S-ELM method.
The training results for the SVM are between
89% and 99% for a range of thresholds. The
corresponding testing results saw gains for some of
the thresholds, while almost same results for the
others. The test results vary from 69% to 89% over a
range of thresholds, for the two class problem. The
results are much better for the S-ELM algorithm,
where the training and testing results are closer
together showing better generalization. The training
results vary between 73 % and 89% while the test
results vary between 71% and 89% which are better
than the results for the SVM and NETASA method.
Our interest in including the SVM in our simulations
was to show the advantages in time factor when the
S-ELM algorithm is used. The training results for
the S-ELM show a little gain over the NETASA and
the SVM results, but the testing results for S-ELM
clearly show higher results of between .006 to 4.476
% as seen in Figure 1 and Table 2. Similarly for the
three-class problem, seen on the last two lines of
Figure 3, the training accuracies for SVM are very
high at 99% while the testing accuracies are 68%
and 54% for two different thresholds, which are
slightly higher than for the NETASA results.
For the S-ELM results, the training accuracies
are closer to the testing accuracies, indicating better
generalization for the 3-class problem also. Here the
S-ELM test results show between 3 to 4% gains as
compared to the NETASA results. As indicated by
many results in the literature, the accuracies can vary
widely for different thresholds and different number
of classes into which the data is divided. A general
trend in the literature is that the RSA prediction
results vary between 70 % and 80%, similar to what
is seen here. So, the S-ELM gives comparable
results to literature.
Table 2: Training and Testing accuracies comparisons
between NETASA, SVM and S_ELM for all thresholds
using 350 hidden neurons are given. The support vectors
are given for SVM data.
NET-
ASA
SVM SVM S-ELM
Thres -
hold %
Accuracy %
SV
Accuracy
for 350
hidden
neurons %
Training
0 89.8 99.9 3837 88.6
5 76.1 99.9 5894 79.8
10 75.2 99.9 6610 74.0
20 73.1 99.9 6826 72.98
50 80.1 99.9 5897 79.80
10 -20 65.1 99.9 7075 67.12
25 -50 60.9 99.9 7087 63.51
Testing
0 87.9 89.1 ** 89.1
5 74.6 76.2 ** 77.3
10 71.2 71.2 ** 73.1
20 70.3 69.5 ** 71.3
50 75.9 76.3 ** 77.3
10-20 63 64.1 ** 66.0
25-50 55 58.1 ** 59.5
Figure 2: Processing time for modelling: SVM Vs. S-
ELM, clear shows huge gains in time for S-ELM.
The biggest advantage of using S-ELM comes
from the speed at which the data can be processed
by the algorithm, while providing us with slightly
better accuracies. It can be clearly seen from Table 3
that S-ELM has a clear advantage when it comes to
processing speed. The same number of samples of
Accuracy - NETASA Vs S-ELM
0
20
40
60
80
100
0% 5% 10% 20% 50% 10-20 % 25-50 %
Threshold
Accuracy %
NETASA S-ELM
AN EXTREME LEARNING MACHINE CLASSIFIER FOR PREDICTION OF RELATIVE SOLVENT
ACCESSIBILITY IN PROTEINS
367

7545 training sample residues was used for model
building for both algorithms. The ratio of time taken
by SVM and S-ELM for model building, for the
various thresholds range from 20.562 : 175 seconds
which amounts to almost 8.51 times time gain by S-
ELM for 0% threshold data. We find that the time
gains range from 8 times to multiple folds, the
highest being for the 20% threshold data where the
ratio is 20.562:1372.2 which is a gain of over 66.734
times. Generally, the time taken for model building
is most crucial, since the model needs to learn as
much as possible in the shortest time.
Figure 3: Processing time for testing: SVM Vs S-ELM.
For real time applications and for batch
processing applications it might be useful to have
faster testing capabilities and here we see that the S-
ELM algorithm is much faster in its testing
capabilities also. The same number of 43137 testing
residues was used here for the test runs in both
algorithms. Here the time gains between the testing
times for 0 % threshold is .922:410 which amounts
to 444.69 times fastr processing by S-ELM. We find
similar gains for other thresholds with the highest
gain for the 20% threshold at .937:857 which is
914.62 times faster processing speed. Both the SVM
and the S-ELM were run on the same computer
running XP windows operating system with 4 GB
RAM and Matlab software.
Time taken for training and testing runs by SVM
and S-ELM algorithms is given in Table 3. Figure 2
and Figure 3 illustrate the high processing time of
SVM and the very low and steady processing times
of S-ELM very clearly. The time taken by S-ELM is
very low at less than one or two seconds, shown as a
horizontal line close to the x-axis while the time
taken by SVM is quite high, ranging between 200
and 1400 seconds for training and between 400 and
900 seconds for testing. S-ELM takes very little time
for testing since stored optimal parameters are used
to calculate the output analytically using ELM.
There is no processing time data available to
compare speeds with the NETASA method. Future
studies will concentrate on increasing the accuracy
of S-ELM further using optimization techniques to
tune the S-ELM parameters for RSA prediction.
Table 3: Processing time for modelling, training and
testing: comparison between SVM and ELM.
SVM S-ELM
Time in Seconds Time in Seconds
Threshold %
Modelling
Training
Testing
Modelling
Training
Testing
0 175 24.6 410 20.6 0.5 0.92
5 990 105 561 20.8 0.6 0.94
10 1273 67 686 20.9 0.6 0.92
20 1372 76 857 20.9 0.5 0.94
50 977 89 645 20.9 0.6 0.95
10-20 1239 88 723 21.0 1.1 1.08
25-50 226 74 728 21.0 0.7 1.08
4 CONCLUSIONS
We have used the SVM and S-ELM methods of
classification for RSA prediction, using the Manesh
data set. We have compared the performance of
these algorithms with each other and with NETASA,
with respect to the speed of processing and have
shown that there are multiple-fold gains in
computational efficiency while using S-ELM
algorithm. It will be advantageous to use the S-ELM
algorithm for real time and batch processing
applications where accuracy and speed are equally
important.
ACKNOWLEDGEMENTS
We acknowledge the support of National Institutes
of Health through grants R01GM081680,
R01GM072014, and R01GM073095 and the support
of the NSF grant through IGERT-0504304.
REFERENCES
Adamczak, R., Porollo, A., & Meller, J. 2005. Combining
prediction of secondary structure and solvent
accessibility in proteins. Proteins, 59(3) 467-475.
Processing Time - Testing
0
100
200
300
400
500
600
700
800
900
0% 5% 10% 20% 50% 10-20
%
25-50
%
Threshold
Time in Seconds
SVM
S- E L M
ICFC 2010 - International Conference on Fuzzy Computation
368

Altschul, S., Madden, T., Schaffer, A., Zhang, J., Zhang,
Z., Miller, W., & Lipman, D., 1997. Gapped BLAST
and PSI-BLAST: a new generation of protein database
search programs. Nucleic Acids Res, 25(17) 3389-
3402.
Berezin, C., Glaser, F., Rosenberg, J., Paz, I., Pupko, T.,
Fariselli, P., Casadio, R., & Ben-Tal, N., 2004.
ConSeq: the identification of functionally and
structurally important residues in protein sequences.
Bioinformatics, 20, (8) 1322-1324.
Bondugula, R. & Xu, D., 2008. Combining Sequence and
Structural Profiles for Protein Solvent Accessibility
Prediction, In Comput Syst Bioinformatics ConfC,
195-202.
Carugo, O., 2003. Prediction of polypeptide fragments
exposed to the solvent. In Silico Biology, 3(4), 417-
428.
Chen, H., Zhou, H.-X., Hu, X., & Yoo, I., 2004
Classification Comparison of Prediction of Solvent
Accessibility from Protein Sequences, In 2nd Asia-
Pacific Bioinformatics Conference (APBC), 333-338.
Cheng, J., Sweredoski, M., & Baldi, P., 2006. DOMpro:
Protein Domain Prediction Using Profiles, Secondary
Structure, Relative Solvent Accessibility, and
Recursive Neural Networks. Data Mining and
Knowledge Discovery, 13(1) 1-10
Cortes, C. & Vapnik, V., 1995. Support vector networks.
Machine Learning, 20, 1-25.
David, M. P., Asprer, J. J., Ibana, J. S., Concepcion, G. P.,
& Padlan, E. A., 2007. A study of the structural
correlates of affinity maturation: Antibody affinity as a
function of chemical interactions, structural plasticity
and stability. Molecular Immunology, 44 (6), 1342-
1351.
Fan, R. E, Chen, P. H. and Lin, C. J., 2005. Working set
selection using second order information for training
SVM. Journal of Machine Learning Research, 6,
1889-1918.
Gianese, G., Bossa, F., & Pascarella, S., 2003.
Improvement in prediction of solvent accessibility by
probability profiles. Protein Engineering Design and
Selection, 16(12) 987-992.
Huang, G. B., Zhu, Q. Y., & Siew, C. K., 2006. Extreme
learning machine: Theory and applications.
Neurocomputing, 70, (1-3) 489-501
Kim, H. & Park, H., 2004. Prediction of protein relative
solvent accessibility with support vector machines and
long-range interaction 3D local descriptor. Proteins -
Structure, Function, and Bioinformatics, 54 (3), 557-
562.
Manesh, N. H., Sadeghi, M., Arab, S., & Movahedi, A. A.
M., 2001. Prediction of protein surface accessibility
with information theory. Proteins - Structure,
Function, and Genetics, 42 (4) 452-459.
Meshkin, A. & Ghafuri, H., 2010. Prediction of Relative
Solvent Accessibility by Support Vector Regression
and Best-First Method. EXCLI, 9, 29-38.
Mucchielli-Giorgi, M. H., Hazout, S., & Tuffery, P., 1999.
PredAcc: prediction of solvent accessibility.
Bioinformatics, 15 (2) 176-177.
Nguyen, M. N. & Rajapakse, J. C., 2005. Prediction of
protein relative solvent accessibility with a two-stage
SVM approach. Proteins, 59, (1) 30-37.
Ooi, T., Oobatake, M., Namethy, G., & Scheraga, H. A.,
1987. Accessible surface areas as a measure of the
thermodynamic parameters of hydration of peptides.
Proceedings of the National Academy of Sciences of
the United States of America, 84 (10) 3086-3090.
Petersen, B., Petersen, T. N., & Andersen , P., Nielsen,
M., Lundegaard, C., 2009. A generic method for
assignment of reliability scores applied to solvent
accessibility predictions. BMC Structural Biology,
9:51.
Pollastri, G., Baldi, P., Fariselli, P., & Casadio, R., 2002.
Prediction of coordination number and relative solvent
accessibility in proteins. Proteins, 47(2) 142-153.
Pollastri, G., Martin, A., Mooney, C., & Vullo, A., 2007.
Accurate prediction of protein secondary structure and
solvent accessibility by consensus combiners of
sequence and structure information. BMC
Bioinformatics, 8, (1) 201
Saraswathi, S., Suresh, S., Sundararajan, N., Zimmerman,
M. and Nilsen-Hamilton, M., 2010. ICGA-PSO-ELM
approach for Accurate Cancer Classification Resulting
in Reduced Gene Sets Involved in Cellular Interface
with the Microenvironment. IEEE Transactions in
Bioinformatics and Computational Biology,
http://www.computer.org/portal/web/csdl/doi/10.1109/
TCBB.2010.13.
Suresh, S., Saraswathi, S., Sundararajan, N., 2010.
Performance Enhancement of Extreme Learning
Machine for Multi-category Sparse Data Classification
Problems. Engineering Applications of Artificial
Intelligence,
http://dx.doi.org/10.1016/j.engappai.2010.06.009.
Shandar, A. & Gromiha, M. M., 2002. NETASA: neural
network based prediction of solvent accessibility.
Bioinformatics, 18(6), 819-824.
Shen, B. & Vihinen, M., 2003. RankViaContact: ranking
and visualization of amino acid contacts.
Bioinformatics, 19(16), 2161-2162.
Sim, J., Kim, S.-Y., & Lee, J., 2005. Prediction of protein
solvent accessibility using fuzzy k -nearest neighbor
method. Bioinformatics, 21(12), 2844-2849.
Singh, Y. H., Gromiha, M. M., Sarai, A., & Ahmad, S.,
2006. Atom-wise statistics and prediction of solvent
accessibility in proteins. Biophysical Chemistry,
124(2), 145-154.
Wagner, M., Adamczak, R., Porollo, A., & Meller, J.,
2005. Linear regression models for solvent
accessibility prediction in proteins. J Comput Biol,
12(3), 355-369.
Wang, J. Y., Lee, H. M., & Ahmad, S., 2007. SVM-
Cabins: prediction of solvent accessibility using
accumulation cutoff set and support vector machine.
Proteins, 68(1), 82-91.
Zarei, R., Arab, S., & Sadeghi, M., 2007. A method for
protein accessibility prediction based on residue types
and conformational states. Computational Biology and
Chemistry, 31(5-6) 384-388.
AN EXTREME LEARNING MACHINE CLASSIFIER FOR PREDICTION OF RELATIVE SOLVENT
ACCESSIBILITY IN PROTEINS
369
