
SEGMENTATION OF SES FOR PROTEIN STRUCTURE
ANALYSIS
Virginio Cantoni, Riccardo Gatti and Luca Lombardi
University of Pavia, dept. of Computer Engineering and Systems Science,Via Ferrata 1, Pavia, Italy
Keywords: Protein structure analysis, Protein-protein interaction, Surface labeling, Convex hull, Distance transform.
Abstract: The morphological complementarities of molecular surfaces provides insights for the identification and
evaluation of binding sites. A quantitative characterization of these sites is an initial step towards protein
based drug design. The final goal of the activity here presented is to provide a method that allows the
identification of sites of possible protein-protein and protein-ligand interaction on the basis of the
geometrical and topological structure of protein surfaces. The goal is to discover complementary regions
(that is with concave and convex segments that match each others) among different molecules. In particular,
we are considering the first step of this process: the segmentation of the protein surface in protuberances and
cavities through an approach based on an analysis of the molecule Convex Hull and on the Distance
Transform.
1 INTRODUCTION
An important research activity, with the large set of
proteins in the current Protein Data Bank (PDB), is
the prediction of interactions of these molecules by
the discovery of similar or of complementary
regions on their surfaces. When a novel protein with
unknown functionalities is discovered,
bioinformatics tools are used to screen huge datasets
of proteins searching for candidates binding sites.
More specifically, if a surface region of the novel
protein is similar to that of the active site of another
protein with known function, the function of the
former protein can be inferred and also its molecular
interaction can be predicted. Active sites are
generally in concave and deep spots of the surface
that are called “pockets”.
Much work has been done on the identification
and the analysis of the binding sites of proteins using
various approaches based on different protein
surface descriptions and matching strategies. The
techniques employed are ranging from geometric
hashing of triangles of points and their associated
physico-chemical properties (Shulman, 2004), to
clustering based on a representation of surfaces in
terms of spherical harmonic coefficients (Glaser,
2006) or by a collection of spin-images (Bock, 2007
– Bock, 2008) or by context shapes (Frome, 2004),
to clique detection on the vertices of the triangulated
solvent-accessible-surface (SAS) (Akatsu, 1996), to
local surface ‘buriedness’ evaluation (Brady, 2000).
The goal of this work is to segment the protein
surface in protuberances and cavities. This
segmentation is based on the Distance Transform
(DT) applied to the volume obtained subtracting the
molecule to the its Convex Hull (CH). Once
obtained protrusions and inlets, for each segment, a
few features are provided including area and volume
of the inlet, area and circumference of the pocked
mouth opening, curvature (Cantoni, 2009) and travel
depth. These features are the basic parameters for
the first screening of compatible sites. A more
precise subsequent experimental analysis on a
limited subset of cases must be then applied.
This paper is organized as follows: section two
shows a survey of approaches for segmentation and
analysis of protein surfaces through the convex hull;
in section three is introduced the solution proposed,
then in section four a few results on artificial test-
images and on true proteins molecules are presented.
The final section, section five, provides a few
concluding remarks and briefly describes our
planned activity in the near future.
83
Cantoni V., Gatti R. and Lombardi L. (2010).
SEGMENTATION OF SES FOR PROTEIN STRUCTURE ANALYSIS.
In Proceedings of the First International Conference on Bioinformatics, pages 83-89
DOI: 10.5220/0002590900830089
Copyright
c
SciTePress

2 SURFACE ANALYSIS
SEGMENTATION THROUGH
THE CONVEX HULL
The CH of a molecule is the smallest convex
polyhedron that contains the molecule points. In R
3
the CH is constituted by a set of facets, that are
triangles, and a set of ridges (boundary elements)
that are edges. A practical O(n log n) algorithm for
general dimensions CH computing, is Quickhull
(Barber, 1996), that uses less space and executes
faster than most of the other algorithms.
The CH approach for molecular segmentation is
not new. The first paper applying this method, to
authors’ knowledge, is (Meier, 1995). The Quickhull
algorithm, is applied to the SAS which is defined by
the center of a water-sized probe sphere (usually
with radius values ranging between 1.4 and 1.8 Å)
‘rolling over the van der Waals surface of the atom’.
The technique is based on two specificities: i) the
tips lie directly on the CH surface: they are the
common points between molecule and CH surfaces;
ii) inlets and holes are ‘normally’ covered by large
facets of the CH surface. Both specificities are not
necessary conditions (the second is even not
sufficient) to establish the existence of true tips and
inlets: it is just a reasonable first hypothesis.
Moreover, the technique for tip segmentation is
based on a heuristic approach: each tip is extended
on the outside facet for a distance determined by a
global parameter.
Two other different approaches based on the CH
of the atoms centers have been proposed
by
Edelsbrunner (Edelsbrummer, 1998) and Xie (Xie,
2007). Both apply the Dealunay triangulation
technique, that in 3D have complex counterparts (3D
tetrahedrons), to evaluate quantitatively some
parameters.
The former, through a dual complex (alpha shape)
analysis, provides a quantitative description of the
microenvironments for protein structure based
design. In particular, volume and area of pockets,
area and circumferences of mouth opening, are
evaluated. For the pockets identification, it is used
the discrete flow method, that is the presence of
Delaunay’s tetrahedra disjointed to the dual
complex. In particular, for segmentation purposes,
some geometrical and topological rules allow the
discrimination between two neighboring tetrahedra,
satisfying the previous constraint. Note that, not all
the inlets are identified as pocket (the one for which
the discrete-flow pours to the outside of the CH).
The latter approach is based on a simplified
description that requires only the Cα atoms to
represent the protein structure in order to speed-up
the computation (making the new representation
“scalable to a large data set … yet robust enough to
handle the intrinsic properties of protein
flexibility”). Moreover, the notion of geometric
potential is introduced: this figure quantitatively
describes the microenvironment on the basis of two
heuristic parameters and allows a fast and effective
discrimination for active sites.
Figure 1: Common 2D representations of surface models
for protein’s molecules: i) in green the van der Waals
surface, directly produced from the atom’s locations
through the van der Waals radii; ii) in red the Solvent-
Excluded Surface SES (also known as the molecular
surface or Connolly surface) generated by the envelope of
a rolling sphere over the van der Waals surface (The
radius of the solvent sphere is usually set to the
approximate radius of a water molecule having a van der
Waals radius of 1.4 Å); iii) in blue the solvent accessible
surface (sometimes called the Lee-Richards molecular
surface) generated by the center of the solvent sphere
rolling over the van der Waals surface; iv) in brown the
convex hull, that coincides also with the SES having a
sphere with an infinite radius.
The CH is also the reference surface for the
molecules analysis based on the ‘travel depth’. The
travel depth parameter (Coleman, 2006 – Giard,
2008), with reference to the SES (see figure 1), is
defined as the shortest path accessible for a solvent
molecule between the protein convex hull and a
given point that belongs to the ‘active’ region of
interest (ROI) delimited by CH and SES. It
represents the physical distance that a ‘sufficiently’
small molecule has to travel to approach a surface
position (the pockets bottom are usually the points
of interest). It is particular the case of tunnels, i.e.
when pockets have no ‘bottom’, in which the
molecule can travel through the entire protein and
the travel depth is delimited by two points belonging
to the CH. In particular (Coleman, 2006) introduced
a technique for computing the travel depth on the
basis of a peculiar distance transform
implementation in the ROI defined above. The
Convex hull
Van der Waals surface
Solvent accessibile
Solvent-excluded
BIOINFORMATICS 2010 - International Conference on Bioinformatics
84

implementation proposed by Giard et al. has the goal
of speed-up the travel depth computation through a
surface-based propagation algorithm that should be
in general faster than the volume-based DT.
3 TUNNELS AND POCKETS
DETECTION
In the discrete space the protein and the CH are
defined in a cubic grid V of dimension L x M x N
voxels. Note that the grid is extended one voxel
beyond the minimum and maximum coordinate of
the SES in each orthogonal direction (in this way
both SES and CH borders are inside the V border).
The voxel resolution adopted is 0.25 Å, so as to be
small enough to ensure that, with the used radii in
biomolecules atoms, any concave depression or
convex protrusion is represented by at least one
voxel.
Let us call R the region between the CH and the
SES (the concavity volume (Borgefors, 1996)), that
is:
(1)
Let us call B
CH
the set border voxels of CH, that is:
(2)
Whererepresent the erosion operator of
mathematical morphology and K the discrete
unitarian sphere (in the discrete space a 3x3x3
cube!).
Within the region R the following propagation is
applied:
A = B
CH
;
N = (A ⊕ K)∩R;
E = N – A;
while E ≠ ∅ do
;
A = N;
N = (A ⊕ K)∩R;
E = N – A;
done
where:
i. A represents the increasing set of voxels
contained in R;
ii. E corresponds to the recruited set of near
neighbors of A contained in R (i.e. the voxels
reached by the last propagation step);
iii.
represents the
minimum value among the distances
in
the near neighbors belonging to D already
defined, incremented by the displacement
between the locations (e, n): that is, if e
and n have a common face
; if e and
n have a common edge
; if e and n
have a common vertex
. In three
dimensions, the total number of the near
neighbor elements of p is 26: six of them
that share one face and have distance equal
to 1 from the voxel p, twelve neighbors that
share only an edge and are at distance
and eight that share only a vertex and are at
distance
always from voxel p. At each
iteration, new voxels, inside R, are reached
by the propagation process and the value
they take is determined by the neighbor
distance (from the convex hull) and the
voxels distance from the neighbor involved;
this in order to simulate an isotropic
propagation process and the proper distance
evaluation.
iv. E = ∅ corresponds to the regime
condition: no other changes are given and
the connected component of R, adjacent to
the border B
CH
,
is completely covered.
The values in D represent the distance of each voxel
of A from the border of B
CH
and A corresponds to
the connected component of R adjacent to the
border.
Having A, it is possible to easily identify and
eventually remove the cavities C, that are the
volumes completely enclosed in the macromolecule
M:
C = CH - A - M (3)
Figure 2a: A 2D example for tunnels and pockets
detection composed of three connected components (in
brown). The closed curve in black corresponds to the
convex hull, and the border BCH in gray embodies the
area under analysis and is the starting set of voxels for the
propagation process.
SEGMENTATION OF SES FOR PROTEIN STRUCTURE ANALYSIS
85

Figure 2b: Results achieved after the propagation phase.
Three pockets are identified, in blue, red and violet
respectively. For the other three sides the propagation
process converges: firstly there is the merging of the
green and yellow waves, then the yellow and brown
waves, and the complete coverage of the accessible areas
is achieved in location L and I respectively. Note the three
internal inaccessible components, in white, with labels
α, β, γ.
Figure 2c: Final segmentation, showing the tunnel (in
green) and the three detected pockets.
Figure 2d: Another representation of the results after the
propagation phase. The letters B, D, F correspond to the
top of three pockets (see also figure 2b)). The letters A, C,
and E correspond to three local tops that are adjacent to
important inlets. The letters g and h corresponds to
convergences towards the tops I and L which identify a
threelobate tunnel.
In order to separate the different pockets and
tunnels the volume A must be partitioned into a set
of disjoint segments P
SES
= {P
1
, … , P
j
, … , P
N
},
where N is the number of inlets. The partition must
satisfy the following condition:
P
i
∩ P
j
=
∅
, i ≠ j
(4)
P
1
∪ ·· · ∪ P
j
∪ ·· · ∪ P
N
= A
(5)
As can be easily extended form the 2D example
of Figure 2, starting from the total set of convex hull
facets, several waves are generated and propagation
proceeds up to the complete coverage of the volume
A: the connected component of R adjacent to the
border. During the propagation phase two sets of
salient points are identified: local tops LT
(represented by capital letters in Figure 2) and wave
convergence WC points (lower case).
The LT set is exploited for the segmentation process.
The cardinality of LT corresponds to N
max
the
maximum number of segments/inlets that can be
considered. The effective number of segments, that
determines obviously the number and the
morphology of pockets and tunnels, is found out on
the basis of two heuristic parameters: i) the
minimum travel depth value of the local tops TD
LT
;
ii) an evaluation of near neighbor pivoting effects
PEs. The threshold TD
LT
is introduced because the
surface’s irregularities and the digitalization process
produce small irrelevant spurious cavities. The
thresholds PEs take into account morphological
aspects insight important cavities and can be
characterized by two different features: the nearness
of others, more significant, local tops (τ
1
) and the
BIOINFORMATICS 2010 - International Conference on Bioinformatics
86

relative values of the local-top travel-distance (τ
2
). in
general faster than the volume-based DT.
4 IMPLEMENTATION AND
RESULTS
We start from the ‘space-filling’ representation of
the protein, where atoms are represented as spheres
with their van der Waals radii (this representation is
directly derived from PDB files which supply the
ordered sequence of 3D positions of each atom’s
center). Figure 3 shows the image produced by our
package for Apostreptavidin Wildtype Core-
Streptavidin with Biotin structure (1MK5 in PDB).
Figure 3: On the left the ‘space filling’ representation of
1MK5. The colors follow the standard CPK scheme. On
the right the correspondent secondary structure
representation.
A first critical decision is the space resolution level
for the analysis. The results presented here are given
with a resolution of 0.25 Å, which entails a van der
Waals radius of more than five voxels to the smallest
represented atoms. The algorithm is then applied to
the SES obtained from the quoted surface, after the
execution of a closure operator using a sphere with
radius of 1.4 Å , about 6 voxels, (corresponding to
the conventional size of a water molecule) as
structural element. It is worth to point out that this
closure operation excludes possible passage through
apertures with a section of less than 6.15 Å
2
(about
99 voxels). Note that, as it has been mentioned in
section 3, the grid is extended one voxel beyond the
minimum and maximum coordinate of the SES in
each orthogonal direction (in this way both SES and
CH borders are inside the V border).
Two other parameters characterize the execution:
the minimum passage section θ
1
(obviously
θ
1
≥100); the maximum mouth aperture θ
2
. The
former has been fixed on a heuristic base to 150
Voxels (which for a circle corresponds to a radius of
7 voxels). The latter has been applied with two
different values: 2000, 7500.
Figure 4: A representation of the results after two
segmentation phases with θ
2
equal to 2000 and 7500 in a)
and b) respectively. Note that no tunnel is present, and 9
pockets have been identified and differently colored. In
particular the first three pockets in the top figure are then
represented in figures 5, 6 and 7 corresponding to θ
2
=2000
and the first two pockets in figures 8 and 9 having
θ
2
=7500.
In Figure 4 the results of the segmentation
process are given. This process is executed in two
phase: in the onward propagation the set of the
pocket’s local top is identified; later a backward
parallel propagation from each of the tops with
identify all the pockets is executed. Finally, it is
applied the near neighbor pivoting process, which
(with τ
1
=10 and τ
2
=5) leads to the final result
shown. Figures 5-9 show the pockets with the
highest travel distance (obviously the parameters of
reference can be one of the others features – e.g. the
pocket volume -, or even a combination of features –
e.g. travel distance and pocket volume-) for different
values of the parameter θ
2
. Each pocket is
represented with a lateral and frontal view
(respectively on the left and on the right side of each
figure).
As it can seen from figure 4, having θ
1
=150 voxels
no tunnels are present, but there are several well
characterized pockets which can be easily
characterized and evaluated. In particular the
dependence of the results of the segmentation
process from the different parameters is pointed out.
The algorithms has been tested on a 2.20 GHz x86
Intel processor the computing time is about 12
seconds for the protein 1MK5 with 127 residues and
1844 atoms.
SEGMENTATION OF SES FOR PROTEIN STRUCTURE ANALYSIS
87
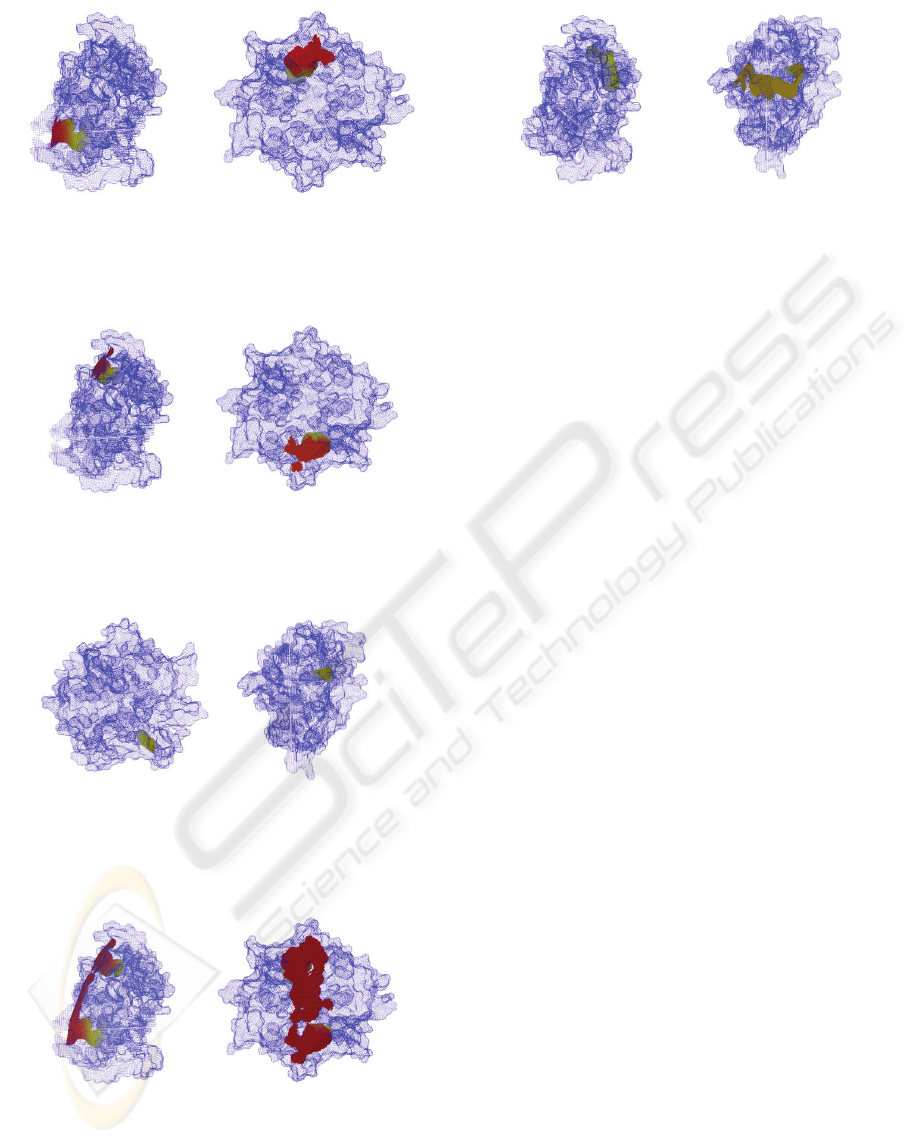
Figure 5: The pocket with the highest travel depth (highest
distant from the CH): 57, with the control parameter θ
2
to
the value: 2000. Note that the red component testifies that
the ‘pocket mouth’ is close to the CH.
Figure 6: The pocket with the second highest travel depth:
35, with θ
2
= 2000. Note that also in this case, the ‘pocket
mouth’ is close to the CH.
Figure 7: The pocket with the third highest travel depth:
34, with θ
2
= 2000. Note the absence of the red
component: the ‘pocket mouth’ is at a distance of 20
voxels from the CH.
Figure 8: The pocket with the highest travel depth
achieved with θ
2
= 7500. Note that for this value of the
control parameter the two highest pocket achieved with θ
2
= 2000 are fused together realizing a large ‘pocket mouth’
more close to the CH.
Figure 9: The pocket with the second highest travel depth
with θ
2
= 7500. Note that this is the evolution of the
previous third pocket and is characterized by a large
‘pocket mouth’ and the presence of the red component
(that is the mouth is closer to the CH).
5 CONCLUSIONS
The final goal of the activity here presented is to
provide a method that allows the identification of
sites of possible protein-protein and protein-ligand
interaction on the basis of the geometrical and
topological structure of protein surfaces. The goal is
then to discover complementary regions (that is with
concave and convex segments that match each
others) among different proteins. In particular, we
are considering the first step of this process: the
segmentation of the protein surface in various
pockets and tunnels. The next step of our activity is
related to the characterization of each of the
extracted segment through morphological and
topological quantitative descriptors (including travel
depth, mouth aperture, curvature, volume) that can
be combined with the local biochemical features
(types of residues and their characteristics) to detect
and specialize the active sites of a protein.
REFERENCES
Akutsu T, 1996. Protein structure alignment using
dynamic programming and iterative improvement. In
IEICE Trans. Inf. and Syst., Vol. E78-D, pp. 1-8.
Barber CB, Dobkin DP, and Huhdanpaa H, 1996. The
Quickhull Algorithm for Convex Hull. In ACM
Transactions on Mathematical Software, Vol. 22, N. 4,
pp. 469-483.
Bock ME, Garutti C, Guerra C, 2007. Spin image profile:
a geometric descriptor for identifying and matching
protein cavities. In Proc. of CSB, San Diego.
Bock ME, Garutti C, Guerra, C, 2008. Cavity detection
and matching for binding site recognition.
In Theoretical Computer Science,
doi:10.1016/j.tcs.2008.08.018.
Borgefors G and Sanniti di Baja G, 1996. Analyzing
BIOINFORMATICS 2010 - International Conference on Bioinformatics
88

Nonconvex 2D and 3D Patterns. In Computer Vision
and image Understanding, vol. 63, N. 1, pp. 145-157.
Brady GP, Stouten PFW, 2000. Fast prediction and
visualization of protein binding pockets with PASS. In
J Comput-Aided Mol Des, 14, pp. 383-401.
Cantoni V, Gatti R, and Lombardi L, 2009. Towards
Protein Interaction Analysis through Surface
Labeling. ICIAP 2009, in press.
Coleman RG, Sharp KA, 2006. Travel Depth, a New
Shape Descriptor for Macromolecules: Application to
Ligand Binding. In J. Mol. Biol., Vol. 362, pp. 441-
458. 1MK5 1A0Q 1ATJ PDBbind, 29 di pg 451,
1H2R
Edelsbrunner H, Liang J and Woodward C, 1998.
Anatomy of protein pockets and cavities: measurement
of binding site geometry and implications for ligand
design. In protein Science 7, pp. 1884-1897.
Frome A, Huber D, Kolluri R, Baulow T and Malik J,
2004. Recognizing Objects in Range Data Using
Regional Point Descriptors. In Computer Vision -
ECCV, pp. 224-237.
Giard J, Rondao Alface Patrice, Macq B, 2008. Fast and
accurate travel depth estimation for protein active site
prediction. In SPIE Electronic Imaging 2008, San
Jose, California USA, San Jose Célifornia, USA,
6812, pp. 0Q-10Q.
Glaser F, Morris RJ, Najmanovich RJ, Laskowski RA and
Thornton JM, 2006. A Method for Localizing Ligand
Binding Pockets in Protein Structures. In PROTEINS:
Structure, Function, and Bioinformatics, 62, pp. 479-
488.
Meier R, Ackermann F, Hermann G, Posch S, and Sagerer
G, 1995. Segmentation of molecular surfaces based on
their convex hull. In Proceedings IEEE International
Conference on Image Processing, pp. 552-554. 2utg
Shulman-Peleg A, Nussinov R and Wolfson H, 2004.
Recognition of Functional Sites in Protein Structures.
In J. Mol. Biol., 339, pp. 607-633.
Xie L and Bourne PE, 2007. A robust and efficient
algorithm for the shape description of protein
structures and its application in predicting ligand
binding sites. In BMC Bioinformatics, 8 (Suppl 4):S9.
SEGMENTATION OF SES FOR PROTEIN STRUCTURE ANALYSIS
89
