
PMSGA: A FAST DNA FRAGMENT ASSEMBLER
Juho M¨akinen, Jorma Tarhio
Department of Computer Science and Engineering, Helsinki University of Technology
P.O. Box 5400, FI-02015 TKK, Finland
Sami Khuri
Department of Computer Science, San Jos´e State University, One Washington Square, San Jos´e, CA 95192-0249, U.S.A.
Keywords:
DNA sequencing, Fragment assembly, Pattern matching, Sequence reconstruction, String graph.
Abstract:
The DNA fragment assembly is an essential step in DNA sequencing projects. Since DNA sequencers output
fragments, the original genome must be reconstructed from these small reads. In this paper, a new fragment
assembly algorithm, Pattern Matching based String Graph Assembler (PMSGA), is presented. The algorithm
uses multipattern matching to detect overlaps and a minimum cost flow algorithm to detect repeats. Special
care was taken to reduce the algorithm’s run time without compromising the quality of the assembly. PMSGA
was compared with well-known fragment assemblers. The algorithm is faster than other assemblers. PMSGA
produced high quality assemblies with prokaryotic data sets. The results for eukaryotic data are comparable
with other assemblers.
1 INTRODUCTION
DNA fragment assembly is a technique that attempts
to reconstruct the original DNA sequence from a large
number of fragments, each several hundred base-pairs
long. The DNA fragment assembly is needed because
current technology, such as gel electrophoresis, can-
not directly and accurately sequence DNA molecules
longer than 1000 bases. However, most genomes are
much longer. For example, a human DNA is about
3.2· 10
9
nucleotides in length and cannot be read at
once. The shotgun sequencing technique was devel-
oped to deal with this limitation. First, the DNA
molecule is amplified, that is, many copies of the
molecule are created. The molecules are then cut at
random sites to obtain fragments that are short enough
to be sequenced directly. The overlapping fragments
are then assembled into a target DNA sequence. Sev-
eral algorithms have emerged to tackle the fragment
assembly problem. Some of the most well-known as-
semblers are PHRAP (Green, 1999), CAP3 (Huang &
Madan, 1999) and EULER (Pevzner & al., 2001).
In this paper, a new fragment assembly algo-
rithm, Pattern Matching based String Graph Assem-
bler (PMSGA), is presented. The algorithm is based
on the overlap-layout-consensus paradigm. Pairwise
overlaps are detected by finding common probes
among fragments. Next, the layout is built using a
minimum cost network flow algorithm. Finally, the
consensus sequence is constructed using q-mers and
position based hashing.
The paper is organized as follows. The details of
the different phases of PMSGA are described in Sec-
tion 2. In Section 3, the experimental results and com-
parisons with other algorithms are presented. Finally,
Section 4 concludes the article with suggestions for
future directions and improvements.
2 THE ALGORITHM
PMSGA is an assembler based on the overlap-layout-
consensus paradigm. In the overlap phase, the task is
to find all possible pairwise overlaps between frag-
ments and their reverse complements. In the lay-
out phase, the order in which the fragments should
be placed in the final assembly is determined. Fi-
nally, in the consensus phase, the aim is to construct
the contigs by finding a consensus of the overlapping
fragments. Next, the three phases of PMSGA are
desribed.
77
Mäkinen J., Tarhio J. and Khuri S. (2010).
PMSGA: A FAST DNA FRAGMENT ASSEMBLER.
In Proceedings of the First International Conference on Bioinformatics, pages 77-82
DOI: 10.5220/0002580800770082
Copyright
c
SciTePress

2.1 The Overlap Phase
The overlap phase is based on the AMASS algorithm
(Kim, 1997). Kim used a multipattern matching al-
gorithm to efficiently find possible pairwise overlaps
between fragments.
PMSGA begins by randomly selecting probes of
constant lengths from fragments and their reverse
complements. Then, a faster multipattern matching
algorithm, called the BG algorithm (Salmela & al.,
2006), is used to find the occurrences of these probes
in the fragments.
The BG algorithm is based on the BNDM algo-
rithm (Navarro & Raffinot, 2000) for a single pattern.
The idea is to construct a generalized pattern that rep-
resents a group of patterns. For example, the group
of patterns, acgt, aacc, and gttt can be represented
by the generalized pattern: [a,g][a,c,t][c,g,t][c,t]. In
the overlap phase, PMSGA uses the BG algorithm to
find the generalized pattern representing overlapping
k-mers instead of single characters of the patterns. If
k = 2 in the example above, the corresponding gener-
alized pattern is given by [ac,aa,gt][cg,ac,tt][gt,cc,tt].
Each occurrence of the generalized pattern is a candi-
date for a real match. BNDM works as a filter and the
candidate matches are checked by an exact method. In
practice, the BG algorithm is very efficient (Salmela
& al., 2006).
To deal with small repeats, the probes with too
many occurrences are filtered out. However, if there
are enough probes with about equal number of occur-
rences, they are left in. This is done to preserve large
repeats, but to filter out small repeats that might cause
false overlaps between fragments.
The probe length (m) is obtained by solving m =
lnF/ln(1− ε), where F is the predefined average
probability of probe occurrence and ε is the average
error rate. In this work, the probe length varies be-
tween 10 and 30 base pairs, and F = 0.4 is used.
The overlap between a pair of fragments i and
j is computed as follows. The common probes of
these fragments are found using the BG algorithm.
Let probes a and b occur in i and j at positions
(pos
a,i
, pos
a, j
) and (pos
b,i
, pos
b, j
). Now, a and b are
said to be a consistent pair, if
|(pos
b,i
− pos
a,i
) − (pos
b, j
− pos
a, j
)| < σ
for a small threshold valueσ. This threshold is needed
to deal with insertion and deletion errors in the frag-
ments. A set of common probe occurrences is a con-
sistent set, if each consecutive pair of probe occur-
rences in the set is a consistent pair.
For each consistent set S
i
that can be constructed
from the probe hits in the overlapping area, a score
is calculated as Score(S
i
) = N
d
W + N
m
, where N
d
is
the number of disjoint probe sets within the consistent
set, W is the distance from the first probe in the set to
the last one and N
m
is the total number of probes in
the set. The consistent set with the largest score is
chosen to represent the overlap, and the length of the
overlap is calculated by using the selected set.
The quality of the detected overlaps is checked as
follows. Let a k-mer represent a sequence of k con-
tigous base pairs. Given a fragment a, occurrences
of all possible k-mers in overlapping areas are deter-
mined. A vectorV
a
of length 4
k
is constructed, where
its elements represent the number of occurrences of a
k-mer. Similarly, a vector V
b
for fragment b is con-
structed.
The average error probabilities for the overlapping
area of fragments a and b, denoted by p
a
and p
b
, are
also calculated. To compute these probabilities, the
PHRED quality scores (Ewing & Green, 1998) for
each fragment are used.
The number of k-mers that occur only in one frag-
ment is calculated as follows
N
miss
=
4
k
∑
i=1
V
a
−V
b
i
(1)
The total number of k-mers, N
tot
, is given by N
tot
=
2(L − k), where L is the length of the overlap being
considered. The number of common k-mers is given
by N
hit
= N
tot
− N
miss
.
The probability of a sequencing error in a k-mer is
given by p
m
= 1−((1− p
a
)(1− p
b
))
k
. The probabil-
ity of observing N
miss
with given error probability, is
given by
P(X ≥ N
miss
) =
N
tot
∑
i=N
miss
N
tot
i
p
i
m
(1− p
m
)
N
tot
−i
(2)
A threshold ξ is set to discard all overlaps, where
P(X ≥ N
miss
) ≤ ξ (3)
Note that P(X ≥ N
miss
) is the incomplete beta func-
tion, I
p
m
(N
miss
,N
hit
+ 1), which can be efficiently ap-
proximated.
To validate overlaps, PMSGA counts the distinct
k-mers, instead of counting errors, as was done by Ke-
cecioglu and Myers (1995). Note that by increasing
the value of k, a greater emphasis is placed on the cor-
rect order of the matching substrings within the over-
lap at the expense of a smaller number of common
k-mers. Fragments that are entirely contained in other
fragments are removed and are used in the final con-
sensus phase.
2.2 The Layout Phase
The layout phase of PMSGA is based on string graphs
(Myers, 2005). The idea is to construct a bidirected
BIOINFORMATICS 2010 - International Conference on Bioinformatics
78
overlap graph describing the overlaps and orientations
between fragments. Then, the fast transitive reduction
algorithm (Myers, 2005) is used to perform the tran-
sitive reduction of the graph.
As the overlap detection based on probe matching
produces somewhat imperfect results, it is important
to deal with possible false overlapsin the graph. Upon
performing the transitive reduction, the count of edges
that could be reduced using the given edge is stored
for each remaining edge. A vertex in the graph is de-
fined to be inconsistent if it has more than one in-edge
or out-edge. Every inconsistent vertex is checked to
see if some edges causing the inconsistency can be
removed. The edge is removed if it was not used to
reduce any other edge in the transitive reduction.
Missing overlaps usually cause the transitive re-
duction to produce two separate chains of vertices
with the same direction between the two vertices. A
chain represents a path of vertices where each vertex
has exactly one in-edge and one out-edge. If the sec-
ond chain contains either no vertices or only one inter-
mediate vertex, it is likely to be caused by a missing
overlap. In such cases, the shorter chain is removed
from the graph.
The probability that a consistent chain of frag-
ments in the graph is traversed once in the reconstruc-
tion is given by
N
k
∆
G
G− ∆
G
N−k
≈
∆N
G
k
k!
e
−∆N
G
(4)
where k is the number of fragments in the chain, G is
the size of the genome in base pairs, ∆ is the length
of the fragment chain in base pairs, and N is the total
number of fragments (Myers, 2005). The probability
that the chain is traversed twice can be constructed
in a similar fashion. An A-statistic for the chain is
computed by taking the logarithm of the ratio of these
probabilities (Myers, 2005): A(∆,k) = ∆N/G− kln2.
Upper and lower bounds for the number of traver-
sals for each edge in the graph can be determined by
setting a threshold on the A-statistic. In this work, the
best results were obtained by using a threshold value
of 5.
The actual number of traversals is obtained by
solving this problem as a minimum cost flow problem
(Myers, 2005). In this work, the cycle canceling al-
gorithm was used (Bang-Jensen & Gutin, 2001). The
cost for each edge was calculated from the amount
of probe hit coverage detected in the overlap phase.
Higher probe hit coverage results in lower cost, and
lower hit coverage increases the cost.
In this work, repeats were not assembled. Conse-
quently, all vertices with traversal count higher than
one were removed.
2.3 The Consensus Phase
The order of the fragments was determined in the
layout phase and the relative overlap length between
fragments was computed in the overlap phase. The
approximate position for each fragment within its
contig can now be determined.
A structure called a consensus graph is used to
efficiently produce the final consensus. The construc-
tion of the consensus graph for a given contig begins
by dividing each fragment into a sequence of n−q+1
q-mers, where n is the length of the fragment. An
edge from q-mer A to B is added only if A and B come
from the same fragment, and B is right next to A in
the fragment. The q-mers are placed into a hash table
and are merged into the same node in the graph only
if their approximate positions are close enough in the
contig.
The pseudocode of the consensus graph construc-
tion algorithm is given in Algorithm 1. The algorithm
Algorithm 1. Algorithm for consensus graph con-
struction. C is a list of fragments in a given contig,
ordered by their approximate left positions. The out-
put of the algorithm is the consensus graph G. T is
the maximum distance for node combination thresh-
old and q is the length of the q-mer.
1: G ← {}
2: c ← 0
3: for each F ∈ C do
4: cpos ← leftPosition[F]
5: last ← NULL
6: c ← c + 1
7: while cpos < length[F] + leftPosition[F] − q
do
8: g ← F[cpos:cpos+q-1]
9: find n where |n.pos-cpos| < T, n.mark 6= c
and n.mer = g
10: if n=NULL then
11: n ← new node(g,cpos)
12: G.addVertex(n)
13: end if
14: if last 6= NULL then
15: e ← G.getEdge(last,n)
16: if e = NULL then e ← G.addEdge(last,n)
endif
17: e.score ← e.score + quality[F][cpos+q-1]
18: end if
19: n.mark ← c
20: last ← n
21: cpos ← cpos + 1
22: end while
23: end for
PMSGA: A FAST DNA FRAGMENT ASSEMBLER
79
efficiently calculates consecutive q-mers (line 8) us-
ing bit shifts. The bf find operation on line 9 can be
efficiently implemented by placing all nodes in a hash
table, where the corresponding q-mer is used as a key.
If no node with required characteristics is found, a
NULL is returned. In computing the score (line 17),
PHRED quality scores (Ewing & Green, 1998), are
taken into consideration.
Next, the path with the largest sum of scores yields
a contig. These contigs are used as new fragments,
and the algorithm is executed once more. The pro-
gram terminates when two consecutive runs produce
similar output results.
3 EXPERIMENTAL RESULTS
PMSGA described in the previous section was tested
under several settings. The data sets used to test
the algorithm were obtained from the NCBI assem-
bly archive (NCBI, 2008). Additionally, artificial
data sets were created using the ReadSim simulator
(Schmid & al., 2008). All results were compared to
PHRAP (Green, 1999) and CAP3 (Huang & Madan,
1999) assemblies. Only data sets with ID 1, 2, and 3
were tested with EULER (Pevzner & al., 2001). The
EULER assembler could not be tested with the larger
data sets, with ID 4 to 8, due to high memory require-
ments (see Tables 2 and 3).
The measuring criteria given in Table 1 were used
to evaluate the performance of the algorithms.
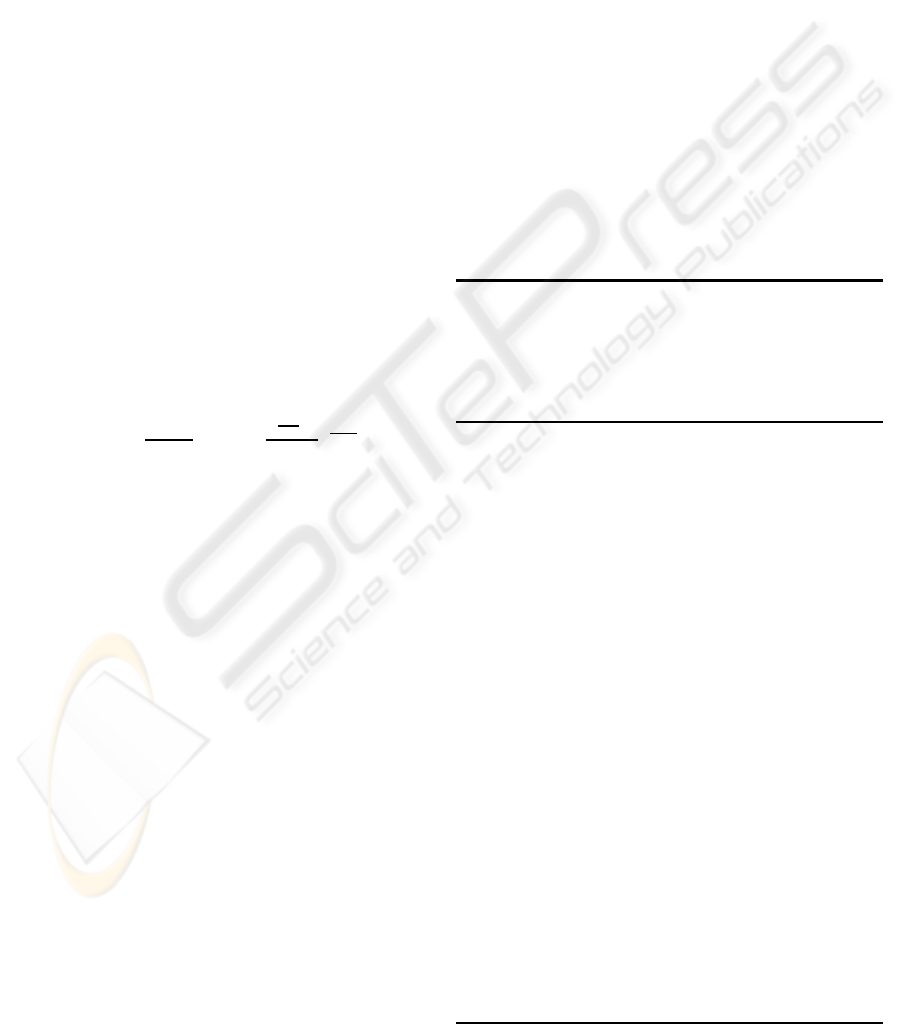
Table 1: The measuring criteria.
Runtime The time to perform a full assembly.
Contigs Number of noncontiguous sequences at
the end of the assembly.
Errors Number of misassembled fragments.
Identity Percentage of identical nucleotides in
the pairwise alignment of the output and
the original sequence.
Sequence Percentage of the original sequence
covered covered by the contigs in the output.
N50 The N50 length is the length x such
that 50% of the sequence is contained
in contigs of length x or greater (Water-
ston & al., 2003).
The first five data sets obtained from the NCBI
assembly archive are described in Table 2. This set
contains three viruses and two bacteria.
The various assemblers were tested with the data
sets of Table 2. The results are reported in Table 3.
As can be seen in Table 3, PMSGA was considerably
faster. PMSGA also produced fewer contigs than the
other assemblers. In 3 out of the 5 cases, PMSGA
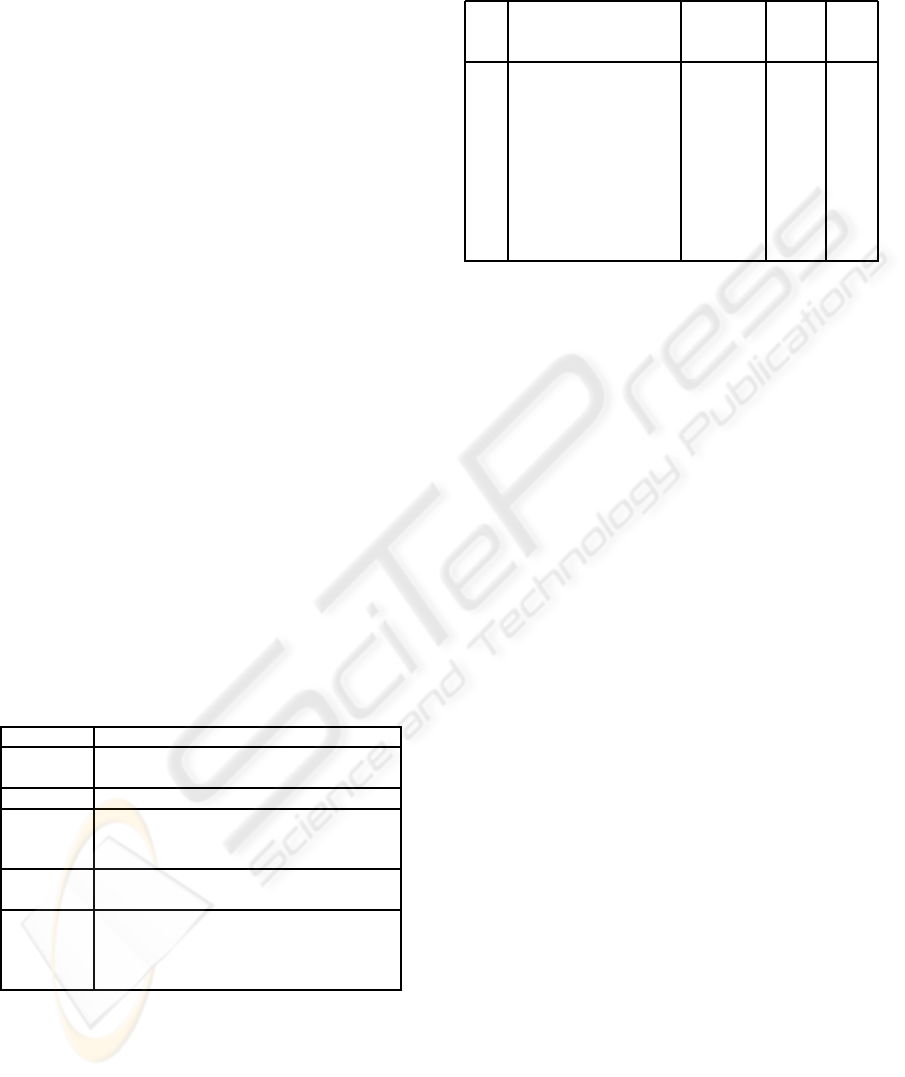
Table 2: Prokaryotic data sets retrieved from NCBI assem-
bly archive (NCBI, 2008).
ID Name Length Seq.
Cov-
ered
Avg.
Er-
ror
1 Antelope corona-
virus US/OH1/2003
31 kbp 15.8 3.1%
2 Bacteriophage
KVP40
240 kbp 10.7 4.1%
3 Chlorella virus
MT325
310 kbp 11.1 1.4%
4 Campylobacter fetus
subsp. fetus 82-40
1.8 Mbp 10.3 1.2%
5 Streptococcus
agalactiae A909
2.1 Mbp 9.0 2.6%
yielded one contig. In test cases with ID 4 and 5, the
output coverage of PMSGA was slightly lower when
compared to the results of CAP3 and PHRAP. This is
probably due to the fact that some fragments that con-
tain repeats were not considered in the final layout.
Next, the assemblers were tested with the eukary-
otic genomes described in Table 5. To create data sets
with ID 6, 7, and 8, the DNA sequences were first
downloaded from the NCBI genome database. Then,
the fragments were created from the sequences, by
using the ReadSim simulator. In these data sets, the
fragment length was uniformly distributed between
600 and 1000 base pairs. Note that PHRED quality
scores are not available for data sets generated with
ReadSim.
The results are reported in Table 4. Once again,
PMSGA outperformed the other assemblers in terms
of speed. PMSGA produced fewer contigs than
CAP3. The number of contigs obtained by PMSGA
is comparable to those obtained by PHRAP. As for
coverage, PMSGA’s results vary between 97.5% and
99.2%.
Next, the percentage of time PMSGA spent on the
four different tasks was measured. The data sets from
Table 2 were used, and the runtimes spent on pattern
matching, overlap list construction, string graph con-
struction and consensus building were recorded. The
results of this experiment are presented in Table 6.
Note that the sum of the percentages reported in Ta-
ble 6 does not add to 100%. The time spent on tasks
such as file I/O and probe selection is not reported.
As can be seen in Table 6, the graph construction
phase is generally the fastest one, followed by pat-
tern matching. Overlap construction is the slowest
phase. The algorithm was implemented in C++. All
tests were run under Linux on AMD Opteron 246/2
GHz dual-processor CPU with 6 gigabytes of mem-
ory. Only one processor was used in testing.
BIOINFORMATICS 2010 - International Conference on Bioinformatics
80
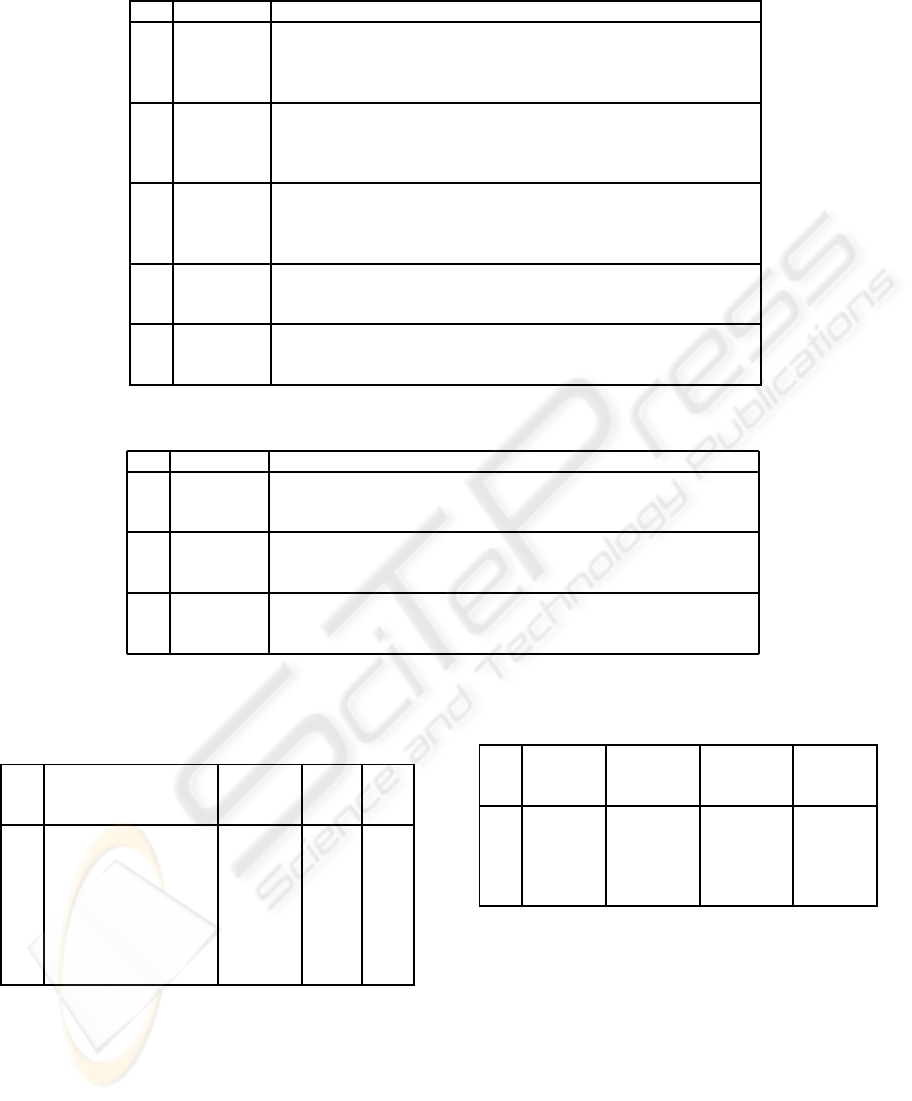
Table 3: Results for assemblies of data sets described in Table 2.
ID Algorithm Runtime Coverage Identity Contigs N50 Errors
1 EULER 5 m 97.8% 98.9% 5 7,676 0
CAP3 1 m 20 s 100% 99.9% 2 19,489 0
PHRAP 11 s 100% 99.8% 5 30,994 0
PMSGA 4 s 100% 100% 1 30,994 0
2 EULER 38 m 61.6% 99.4% 87 6,517 0
CAP3 8 m 20 s 100% 99.4% 118 3,679 0
PHRAP 1 m 10 s 100% 99.8% 16 94,898 0
PMSGA 27 s 100% 100% 1 244,834 0
3 EULER 39 m 99.7% 99.6% 4 256,318 1
CAP3 11 m 100% 99.9% 24 22,941 1
PHRAP 1 m 30 s 100% 100% 1 314,327 0
PMSGA 20 s 99.5% 100% 1 312,631 0
4 CAP3 1 h 40 m 99.2% 99.9% 156 18,101 5
PHRAP 13 m 99.5% 100% 19 218,453 8
PMSGA 2 m 30 s 96.3% 100% 12 619,222 4
5 CAP3 2 h 2 m 99.1% 99.0% 1879 1,868 6
PHRAP 20 m 98.9% 99.9% 399 39,775 7
PMSGA 8 m 30 s 91.1% 99.9% 47 87,267 3
Table 4: Results for assemblies of data sets described in Table 5.
ID Algorithm Runtime Coverage Identity Contigs N50 Errors
6 CAP3 40 m 100% 100% 57 77,313 6
PHRAP 7 m 100% 100% 14 281,034 2
PMSGA 2m 50 s 99.2% 100% 15 187,017 2
7 CAP3 57 m 99.9% 100% 56 103,076 5
PHRAP 11 m 99.8% 100% 27 178,492 3
PMSGA 4 m 20 s 97.9% 100% 34 147,755 3
8 CAP3 1 h 20 m 99.7% 100% 190 54,555 5
PHRAP 15 m 100% 100% 79 117,652 7
PMSGA 8 m 97.5% 100% 95 97,404 7
Table 5: Eukaryotic data sets generated using ReadSim sim-
ulator (Schmid & al., 2008).
ID Name Length Seq.
Cov-
ered
Avg.
Er-
ror
6 Theileria parva
strain muguga
Chromosome 1
2.5 Mbp 8.0 3.0%
7 Pichia stipitis
Chromosome 1
3.5 Mbp 8.0 2.0%
8 Schizosaccharomyces
pombe
Chromosome 1
5.6 Mbp 7.0 2.0%
4 CONCLUSIONS
This article introduced a fast and efficient DNA frag-
ment assembler, PMSGA. PMSGA is faster than
other commonly used assemblers such as PHRAP,
CAP3, and EULER. PMSGA’s speed is due to the
BG algorithm for pattern matching used in the over-
Table 6: Distribution of time spent on the 4 tasks.
ID Pattern
match-
ing
Overlap
construc-
tion
Graph
construc-
tion
Consen-
sus
1 20% 46% 4% 23%
2 15% 51% 8% 23%
3 19% 44% 6% 26%
4 18% 39% 15% 33%
5 6% 44% 16% 33%
lap phase and the algorithm for the consensus graph
construction. In terms of assembly quality, the num-
ber of contigs and the coverage produced by PMSGA
are either better or comparable to the results obtained
by PHRAP.
The experimental results of this work demon-
strated that PMSGA is successful in assembling
prokaryotic genomes as well as eukaryotic genomes.
We are aware that next-generation sequencing
methods are lurking on the horizon. We are aware that
non-Sanger-based sequencing technologies, such as
the ones being developed by Solexa/Illumina, Agen-
PMSGA: A FAST DNA FRAGMENT ASSEMBLER
81

court/ABI, and Helicos Biosciences, are delivering on
their promise of sequencing DNA at unprecedented
speed and that one day they will enable impressive
scientific achievements and novel biological applica-
tions. But as long as Sanger-based sequencing meth-
ods are being used, the DNA fragmentassembly prob-
lem addressed in this work, remains a current and
challenging problem that requires efficient algorithms
such as PSMGA.
PMSGA can further be improved by taking steps
to handle long repeats. Another option would be to
let the researcher manually perform additional tests to
decide the final path in the graph, as suggested by My-
ers (2005). A further improvement could be achieved
by parallelizing the overlap phase.
ACKNOWLEDGEMENTS
The work was financially supported by the Academy
of Finland.
REFERENCES
Green, P. (1999). “Phrap Documentation”. Available:
http://www.phrap.org/phredphrap/phrap.html Refer-
enced June 2009.
Bang-Jensen, J. & Gutin, G. (2001). Digraphs: Theory, Al-
gorithms and Applications, Springer Verlag, 2001.
Ewing, B. & Green, P. (1998). “Base-calling of automated
sequencer traces using Phred. II. Error probabilities,”
Genome Research, vol. 8, no. 3, pp. 186.
Huang, X. & Madan, A. (1999). “CAP3: A DNA sequence
assembly program”, Genome research, vol. 9, no. 9,
pp. 868.
Kececioglu, J. & Myers, E. (1995).“Combinatorial algo-
rithms for DNA sequence assembly,” Algorithmica,
vol. 13, no. 1, pp. 7–51.
Kim, S. (1997). “A structured pattern matching approach
to shotgun sequence assembly,” Ph.D Dissertation,
Computer Science Department, The University of
Iowa, Iowa City.
Salmela, L., Tarhio, J., & Kyt¨ojoki, J. (2006). “Multi-
pattern string matching with q-grams,” ACM Journal
of Experimental Algorithmics, vol. 11, no. 1.
Myers, E. W. (2005). “The fragment assembly string
graph,” Bioinformatics, vol. 21, no. 2.
Navarro, G. & Raffinot, M. (2000). “Fast and flexible string
matching by combining bit-parallelism and suffix au-
tomata, ACM Journal of Experimental Algorithmics,
vol. 5, no. 4.
http://www.ncbi.nlm.nih.gov/Traces/assembly/ Referenced
June 2009.
Pevzner, P. A., Tang, H., & Waterman, M. S. (2001). “An
Eulerian path approach to DNA fragment assembly,”
Proc. Natl. Acad. Sci. USA, vol. 98, no. 17, pp. 9748–
9753.
Schmid, R., Schuster, S. C., Steel, M. A., & Hu-
son, D. H. (2008). “ReadSim – A simulator for
Sanger and 454 sequencing,” in preparation, soft-
ware freely available from www-ab.informatik.uni-
tuebingen.de/software/readsim Referenced June 2009.
Waterston, R., Lander, E., & Sulston, J. (2003). “More on
the sequencing of the human genome,” PNAS, vol.
100, no. 6, pp. 3022–3024.
BIOINFORMATICS 2010 - International Conference on Bioinformatics
82
