
A KDD APPROACH FOR DESIGNING FILTERING
STRATEGIES TO IMPROVE VIRTUAL SCREENING
Leo Ghemtio, Malika Smaïl-Tabbone, Marie-Dominique Devignes, Michel Souchet
and Bernard Maigret
LORIA UMR 7503, CNRS, Nancy-Université and INRIA Research Centre Nancy Grand-Est
BP239, 54506 Vandoeuvre-les-Nancy cedex, France
Keywords: KDD, Heterogeneous data integration, Data retrieval, Data mining, Protein-ligand interaction, 3D structure,
Virtual screening.
Abstract: Virtual screening has become an essential step in the early drug discovery process. Generally speaking, it
consists in using computational techniques for selecting compounds from chemical libraries in order to
identify drug-like molecules acting on a biological target of therapeutic interest. In the present study we
consider virtual screening as a particular form of the KDD (Knowledge Discovery from Databases)
approach. The knowledge to be discovered concerns the way a compound can be considered as a consistent
ligand for a given target. The data from which this knowledge has to be discovered derive from diverse
sources such as chemical, structural, and biological data related to ligands and their cognate targets. More
precisely, we aim to extract filters from chemical libraries and protein-ligand interactions. In this context,
the three basic steps of a KDD process have to be implemented. Firstly, a model-driven data integration step
is applied to appropriate heterogeneous data found in public databases. This facilitates subsequent extraction
of various datasets for mining. In a second step, mining algorithms are applied to the datasets and finally the
most accurate knowledge units are eventually proposed as new filters. We present here this KDD approach
and the experimental results we obtained with a set of ligands of the hormone receptor LXR.
1 INTRODUCTION
In silico drug discovery covers diverse
computational techniques for capturing, integrating
and analyzing biological and chemical data from
diverse sources. Many programs address the issue of
identifying drug-like molecules by calculating the
docking energies of ligands bound to biological
targets. Indeed, virtual screening is recognized today
as a very promising process in early drug discovery
process because it provides an excellent cost-to-
efficiency ratio (Jorgensen 2004; Köppen 2009).
However high-throughput virtual screening methods
are still under-exploited due to the computing cost of
the current docking programs. One way to overcome
such limitations is to couple multiple techniques in a
funnel-like filtering process in which fast selection
methods are used first for discriminating candidates
that can be quickly recognized as consistent for
being passed to the next step of the funnel. Filters
that can be used for this first fast selection step are
classically grouped into two categories. On one
hand, the structure-based methods involve
computing either geometrical matching between
target and ligand, or a combination of features
characterizing the binding mode of ligand to target
(pharmacophore, Finn et al. 1998). These methods
require that the 3D structure of the target is known.
On the other hand, the ligand-based methods rely on
a representative set of reference structures, known to
be biologically active on the target, and compute
structure-activity relationships based on various
molecular descriptors. Both categories (structure-
based and ligand-based) of methods result in a
ranked list of screened compounds.
Actually, the design of a virtual screening filter
can be considered as a particular case of the KDD
(Knowledge Discovery from Databases) approach.
The knowledge to be discovered concerns the
discrimination between good and bad ligands for a
given target, i.e. a classification problem. The data
to be mined for knowledge extraction are chemical,
structural and biological data related to the ligands
and their cognate targets. Indeed the powerful KDD
146
Ghemtio L., Smaïl-Tabbone M., Devignes M., Souchet M. and Maigret B. (2009).
A KDD APPROACH FOR DESIGNING FILTERING STRATEGIES TO IMPROVE VIRTUAL SCREENING.
In Proceedings of the International Conference on Knowledge Discovery and Information Retrieval, pages 146-151
DOI: 10.5220/0002292301460151
Copyright
c
SciTePress

paradigm (Fayyad et al. 1996) provides a consistent
way to address a virtual screening issue. It stresses
the importance of data integration as a first
preparation step and allows diverse mining
algorithms to be applied on several selected subsets
of the integrated data. Knowledge units can be
extracted from these datasets to derive activity
prediction models. Once validated, such prediction
models can be used as a novel type of virtual
screening filters. Since they are produced along a
KDD process, these filters will be called here
“knowledge-based” filters.
The KDD approach presented in this paper
concerns the definition of new virtual screening
filters in a drug discovery context. Special emphasis
is brought to the data integration step since the
ligand descriptor space is huge and complex.
Current programs are able to rapidly calculate
hundreds of molecular descriptors corresponding to
1D, 2D and 3D physico-chemical descriptors. In
most data analysis contexts, data integration efforts
yield a simple matrix of data because most data
mining algorithms accept as input unique tables
where the data are represented as objects displaying
specific values for given properties. However, a
single table representation hardly reflects the
complexity of biological and chemical data related
to Protein-Ligand Interaction (PLI) data. Our
approach is thus rather based on an entity-
relationship data model. An integrated database is
then produced from which various sets of data can
be easily extracted for mining as in Karp et al.
(2008). Interestingly, this architecture revealed to be
useful for solving the multiple-instance learning
problem that arises when considering simultaneously
the descriptors of the ligands and their 3D
conformations.
The proposed KDD methodology has been tested
on three targets corresponding to three distinct 3D
conformers of the same protein. The challenge
addressed here is to combine various sets of ligand
descriptors, pertaining to both structure-based and
ligand-based methods. Section 2 describes the
biological background of this study; the proposed
KDD approach is presented in section 3; section 4
reports on the results of the conducted experiment.
The last section concludes on the advantages and
perspectives of this approach.
2 PREDICTION OF LIGAND
ACTIVITY FOR DRUG
DISCOVERY
Several programs exist for both ligand- and
structure-based screening methods (Kirchmair et al.
2008) and recent developments confirm that
combining results from different methods leads to
better docking performance (Feher 2006). Several
combination methods have been proposed among
which the recent VSM-G approach that designs the
hit identification process as a funnel of several
progressive screening programs (Beautrait et al.
2008). It is composed of a rigid geometrical docking
program (SHEF, Cai et al. 2008) followed by a
flexible docking program (GOLD, Jones et al.,
1997), both programs acting obviously as structure-
based filters. Since the number of false positive hits
is still very high, one direction for improving VSM-
G is to develop knowledge-based filters. This should
reduce the number of false positive hits that are
finally retained.
The KDD approach presented in this paper is
tested with a collection of molecules known for their
activity towards a particular biological target, the
Liver X Receptor (LXR). The LXR receptor is an
attractive target for the development of new
therapeutic agents in the treatment of
cardiovascular-related diseases (Lala, 2005). Reports
on structural characterization of the LXR receptor
reveal a great plasticity of the ligand binding pocket,
which is able to accommodate ligands with different
shapes and sizes (Farnegardh et al. 2003). We
consider in this study three distinct 3D
conformations of the LXR target (codes: 1P8D,
1PQ6, and 1PQ9) obtained by X-ray
crystallography.
3 MODEL-DRIVEN DATA
INTEGRATION AND MINING
Our methodology is composed of four main steps:
(i) building a data model for PLI data taking into
account user requirements and existing resources;
(ii) specifying a workflow for collecting data from
the different resources leading to the specification of
specific wrappers for populating a relational DB;
(iii) writing queries on the data model for each
identified user requirement; (iv) applying a mining
program to the retrieved dataset. The last two steps
can be iterated upon analysis of the extracted
knowledge units.
Figure 1 presents the entity-relationship data
model of the PLI database. The model contains five
entities namely Protein, Ligand, P_L_Interaction,
Protein_Conformer, and Ligand_Conformer,
connected with relevant relationships. A protein is
described by several attributes (e.g. Name,
A KDD APPROACH FOR DESIGNING FILTERING STRATEGIES TO IMPROVE VIRTUAL SCREENING
147
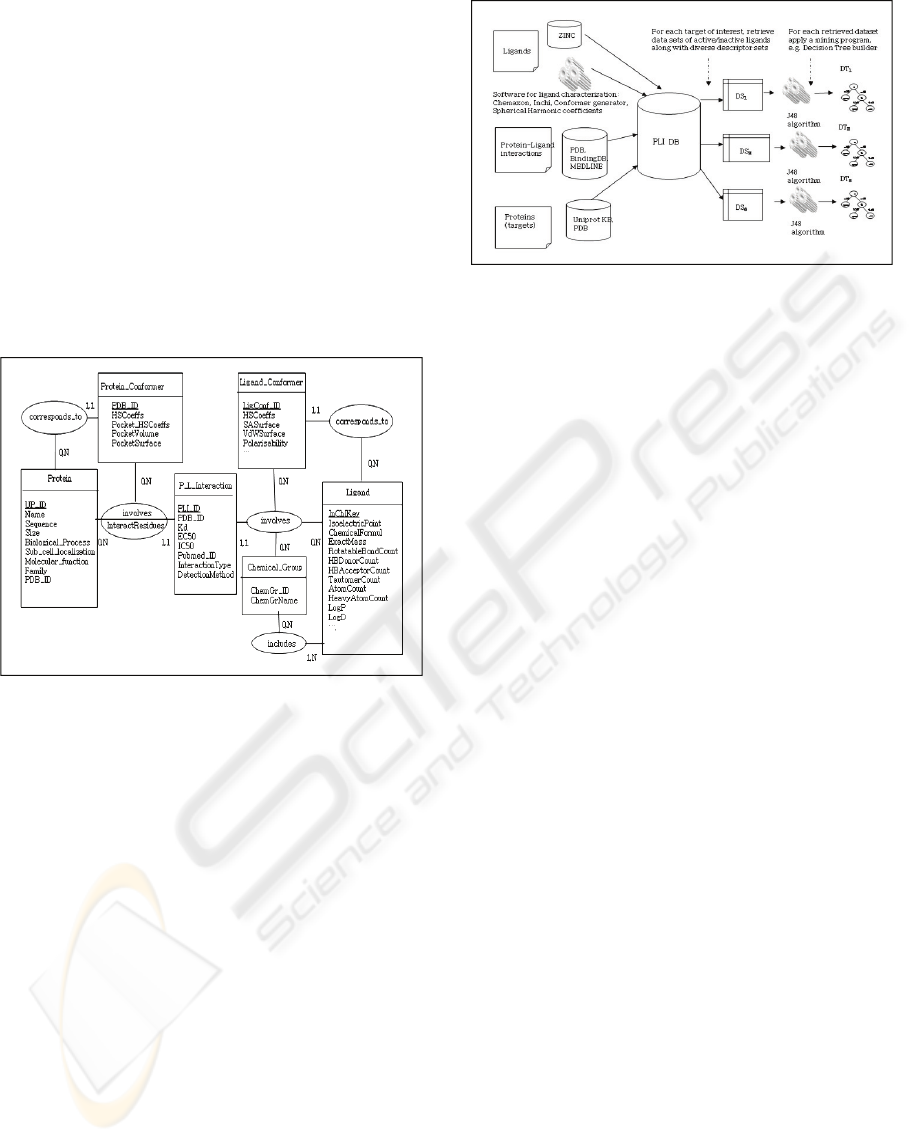
Sequence, Size) available in the UniProtKB protein
knowledge base. A set of physical and chemical
attributes are computed by specific programs from
each ligand with respect to its chemical formula. A
protein may have a known interaction with a ligand.
Each PLI is documented either in the PDB (Protein
Data Bank; Berman et al. 2000), Pubmed or IntAct
databases by a set of characteristics (e.g. EC50, Kd).
The Protein_Conformer and Ligand_Conformer
entities contain topological descriptors of 3D
conformations for each protein and each ligand.
These include Spherical Harmonic (SH) coefficients
which describe the shapes of the target binding site
and of the ligand for easy comparison (Cai et al.,
2008).
Figure 1: The entity-relationship model of the PLI
database.
The overall KDD strategy is figured out in Figure 2.
On the left, the original resources for the data
relative to PLI are represented together with the
main data flows for collecting relevant data
concerning a list of targets of interest and a list of
drug-like molecules. This leads to instanciate the
PLI database (Figure 2, centre) for a given virtual
screening problem.
Once the PLI database is ready, the users can
retrieve various datasets in order to design
knowledge-based filters for virtual screening (Figure
2, right). At this stage of the work, the SQL view
definition mechanism may constitute a powerful
way for retrieving data sets to be mined. A typical
dataset is composed of various ligands (set of
objects) with their values for different descriptors
(set of attributes), including a class attribute (active /
inactive, or binding / not binding). Mining
algorithms can then exploit such datasets in order to
produce prediction models such as decision trees
(DTs). Interestingly, the KDD process adapted to the
Figure 2: KDD process for designing knowledge-based
filters in a virtual screening context.
virtual screening problem facilitates the exploration
of the ligand descriptor space by selecting various
descriptor sets and by evaluating the quality of the
subsequent prediction models.
4 EXPERIMENTAL RESULTS
4.1 Instanciating the PLI Database
The PLI database was constructed according to a
relational data model straight derived from the
entity-relationship model shown in Figure 1. Data
related to the three LXR conformers were imported
from the PDB entries named 1P8D, 1PQ6 and 1PQ9
and used to fill the Protein_Conformer table. In
particular, the structural descriptors of the binding
pocket of each LXR conformer, including their SH
coefficients, were computed and inserted in this
table. A total of 222 LXR ligands were retrieved
from the literature (Spencer et al., 2001; Bennett et
al., 2008; Janowski et al., 1999) and inserted in the
Ligand table. Their activity towards the LXR target
was stored in the Protein_Ligand_Interaction table.
The distinction between active and inactive ligands
was based here on the transactivation (EC50) value
found in the papers cited above. It was arbitrarily
assumed that an active molecule is any molecule for
which the transactivation value has been found
lower than a given threshold of 1μM (micromole per
liter). This criterion yielded 157 active versus 65
inactive ligands in the database. About 20 possible
conformers were generated for each ligand by a
specific program (OpenEye Suite) in order to fill the
Ligand_Conformer table with computed 3D
structural descriptors of ligand conformers.
KDIR 2009 - International Conference on Knowledge Discovery and Information Retrieval
148

4.2 Datasets
Three sets of descriptors were considered for all the
222 ligands from the database. (i) The SAR
descriptor set includes the classical ligand
descriptors used for Structure-Activity Relationship
analysis (Winkler 2002). This set corresponds to
twenty-two attributes of the Ligand table. (ii) The
CONF descriptor set includes six attributes
corresponding to six 3D structural descriptors of
ligand conformers stored in the Ligand_Conformer
table. (iii) The SAR-CONF descriptor set is the union
of the SAR and CONF descriptor sets (28 attributes).
A class attribute (active/inactive) is added to each
descriptor set.
In the CONF and SAR-CONF datasets several
3D conformers are associated with the same ligand.
This leads to a multiple-instance learning problem
(Maron & Lozano-Perez 1998) since the ligand
conformers can be considered as distinct instances of
the ligand, sharing common ligand properties (SAR
descriptors and activity) but having specific
conformer descriptors (CONF descriptors). To solve
this problem we decided to select for each ligand the
best-matching conformer towards each of the three
LXR target conformers. This selection was based on
the highest similarity score calculated with the
SHEF program (Cai et al. 2008) between the SH
coefficients of the ligand conformer on one hand,
and of the binding pocket of the LXR conformer on
the other hand. Finally three single-instance CONF
(respectively SAR-CONF) descriptor sets were
obtained, one for each LXR conformer.
4.3 Construction of Decision Trees
The mining experiments reported in this paper were
carried out with the Weka machine learning program
(Witten & Frank 2005) which includes an
implementation of the J48 version of the C4.5
program for building Decision Trees (DTs) relying
on the divide and conquer principle. The J48
program was run with the default parameters. The
DTs were evaluated by a 10-fold stratified
validation.
There are at least two reasons for using a DT-
type mining algorithm for this experimentation.
Firstly, we want to produce explicit activity
prediction models in which the discriminative
descriptors are made available to the domain
experts. Secondly, the values taken by the
descriptors in the datasets are not binary but rather
numeric, which excludes in a first approach any
symbolic data mining algorithm such as those
searching for frequent itemsets or association rules.
4.4 Evaluation of the Prediction
Models and Discussion
Applying the J48 program on the SAR and SAR-
CONF datasets, using as class attribute the
active/inactive attribute defined in section 4.1, failed
to produce any consistent DT (no descriptor in the
DT). The results simply lead to predicting the major
class in all cases, resulting in an estimation of the
maximal percentage of incorrectly classified
instances of 32 %. Conversely, DTs were obtained
with the CONF datasets for each LXR conformer
using the same active/inactive class attribute. The
observed performances are presented in Table 1.
Table 1: Performance of the DTs predicting the activity of
a ligand conformer with each LXR conformer. FN: False
Negative; FP: False Positive; TP: True Positive.
DT
CONF
LXR conformer 1P8D 1PQ6 1PQ9
#descriptors in the DT 2 2 4
#FN / #FP 4 / 65 6 / 61 2 / 65
% incorrectly classified
instances
31% 30% 30%
Weighted average of TP
rates
0.69 0.7 0.7
The accuracy of the prediction is very low for
the three DTs. About 30% of the instances are
incorrectly classified, which is very close to the
maximal percentage of incorrectly classified
instances. The number of false positive instances is
high (61 to 65). Obviously, these results show that
the considered descriptor sets cannot accurately
predict ligand activity towards any of the three LXR
conformers.
Since it is generally assumed that the activity is
related to the binding, we decided to explore the
capacity of the various descriptor sets to
discriminate between binding and not binding
ligands. Indeed the ultimate screening filter used in
the VSM-G funnel is a flexible docking program
that evaluates a docking score taking into account
the flexibility of both target and ligand conformers.
This flexible docking step requires powerful
computing capacities to be conducted on large sets
of molecules (about one hour is required on one
processor core for docking one molecule on one
target which means about 3 days for one thousand of
molecules on a cluster of 16 bi-quad nodes). We
therefore used the same datasets and simply
A KDD APPROACH FOR DESIGNING FILTERING STRATEGIES TO IMPROVE VIRTUAL SCREENING
149

Table 2: Performance of the DTs predicting the docking of a ligand conformer with a given LXR conformer. Abbreviations
are the same as in Table 1.
DT
SAR
DT
CONF
DT
SAR-CONF
LXR conformer 1P8D 1PQ6 1PQ9 1P8D 1PQ6 1PQ9 1P8D 1PQ6 1PQ9
#docking / #not docking (in the
dataset)
201/21 184/38 115/106 201/21 184/38 115/106 201/21 184/38 115/106
#descriptors (in the dataset) 22 22 22 6 6 6 28 28 28
#descriptors in the DT 1 7 7 1 2 4 1 6 5
#FN / #FP 9 / 9 6 / 11 8 / 13 3 / 12 7 / 16 7 / 13 6 / 9 9 / 11 7/ 13
Incorrectly classified instances 8% 7.6% 9.5% 6.7% 10% 9% 6.7% 9% 9%
Weighted average of TP rates 0.9 0.92 0.91 0.93 0.90 0.91 0.93 0.91 0.91
replaced the active/inactive class attribute with a
docking/not docking class attribute. This
information was produced for each ligand towards
each LXR conformer with the Glide software
(Halgren et al. 2004). The docking score was
converted to a binary class attribute based on a
docking score threshold.
The results are summarized in Table 2. The
DT
SAR
, DT
CONF
, and DT
SAR-CONF
decision trees
correspond to the SAR, CONF, and SAR-CONF
description sets respectively. It appears clearly that
the accuracy of docking prediction is globally much
more satisfying than the accuracy of activity
prediction was (Table 1). Less than 10% of the
instances are incorrectly classified and the number
of false positives is much lower (9 to 16). The
accuracy figures of the DTs obtained with the three
types of descriptor sets towards the three LXR
conformers are very close one to the other. A
possible comparison criterion is the number of
attributes used in each DT, assuming that more
efficient DTs use less attributes for the same
accuracy. With such an hypothesis, the DTs
CONF
perform better than the DTs
SAR
and DTs
SAR-CONF
for
the three LXR conformers. For illustration, Figure 3
shows the docking DT
CONF
obtained for the 1PQ6
LXR conformer. The contribution of all these
suggested filters has now to be evaluated upon
screening a large molecule database against the
considered targets. In particular it will be interesting
to compare the efficiency of the VSM-G screening
funnel with and without these additional filters.
The discrepancy observed between the activity
and the docking prediction models raises the
question of the differences that exist between
binding and activity. Indeed, a retrospective analysis
of the 222 molecules of our dataset reveals that for
each target conformer (i) some active ligands are
found unable to dock and (ii) some inactive ligands
DreidingEnergy<=239:yes
DreidingEnergy>239
|VdWSurface<=705:yes
|VdWSurface>705:no
Figure 3: Docking DT
CONF
for the 1PQ6 LXR conformer.
are docked. This apparent paradox can be explained
by the fact that activity information is captured from
functional biological tests in which the protein can
adopt different conformations in addition to the three
ones tested in the present study. Moreover,
functional tests are designed for active compounds
and cannot distinguish between binding and not
binding inactive compounds.
5 CONCLUSIONS AND
PERSPECTIVES
Our methodology for data integration and mining
includes the rigorous construction of an integrated
database in which data are collected from various
resources. Careful design of such a database
facilitates data preparation and selection upstream
various data mining procedures when searching for
significant hidden patterns. Moreover, it may help
solving the multiple-instance learning problem by
providing rapid access to the information required
for converting a dataset into a single-instance one.
We have illustrated our approach with PLI data
in a specific context of drug discovery. We have
shown how the KDD methodology enables an actual
exploratory data mining approach, leading to the
choice of the best prediction models given three
KDIR 2009 - International Conference on Knowledge Discovery and Information Retrieval
150

types of descriptor sets. In our case, the suggested
KDD approach succeeded in unifying the ligand-
and structure-based approaches for virtual screening.
The prediction models based on the CONF
descriptor set can now be tested as knowledge-based
filters in the VSM-G screening funnel upstream the
flexible docking step in order to reduce the number
of molecules to test with the docking software.
We see two main directions for future work.
Firstly, we plan to use relational data mining
methods for mining relational data and producing
more expressive regularities (Finn et al., 1998;
Dzeroski & Lavrac, 2001; Page & Craven, 2003).
This would allow taking into account the chemical
groups composing a ligand as well as atom-specific
attributes. Secondly, we want to explore various
definitions of ligand activity together with sets of
relational descriptors for producing improved
activity prediction models.
REFERENCES
Beautrait, A. et al. 2008. Multiple-step virtual screening
using VSM-G: overview and validation of fast
geometrical matching enrichment, Journal of
Molecular Modeling, 14, 135-48.
Bennett, D.J., Carswell, E.L., Cooke, A.J., Edwards, A.S.
& Nimz, O. 2008. Design, structure activity
relationships and X-Ray co-crystallography of non-
steroidal LXR agonists. Curr Med Chem 15, 195-209.
Berman, H., WestBrook, J., Feng, A., Gililand, G., Bhat,
T., Weissig, H., Shinlyalov, I., Bourne, P., 2000. The
Protein Data Bank. Nucl. Acid. Res. 28: 235-242.
Cai, W., Xu J., Shao X., Leroux V., Beautrait A., Maigret
B., 2008. SHEF: a vHTS geometrical filter using
coefficients of spherical harmonic molecular surfaces.
J Mol Model 14, 393-401.
Dzeroski, S., and Lavrac, N.(Eds.), 2001. Relational Data
Mining. Springer.
Fayyad, U., Piatetsky-Shapiro, G., Smyth, P., 1996. From
Data Mining to Knowledge Discovery: an Overview.
MIT Press, Cambridge MA.
Feher, M. (2006) Consensus scoring for protein-ligand
interactions, Drug Discovery Today, 11, 421-428.
Finn, P., Muggleton, S., Page, D., Srinivasan, A., 1998.
Pharmacophore Discovery Using the Inductive Logic
Programming System PROGOL. Machine Learning 30(2-
3): 241-270.
Halgren, T. A., Murphy, R. B., Friesner, R. A., Beard, H.
S., Frye, L. L., Pollard, W. T., Banks, J. L. 2004.
Glide: A New Approach for Rapid, Accurate Docking
and Scoring. J. Med. Chem., 47, 1750-1759.
Janowski, B.A. et al. 1999. Structural requirements of
ligands for the oxysterol liver X receptors LXRalpha
and LXRbeta. Proc Natl Acad Sci U S A 96, 266-71.
Jones G., Willett P., Glen R.C., Leach A.R., Taylor R.
1997. Development and validation of a genetic
algorithm for flexible docking. J Mol Biol., 267, 727-
48.
Jorgensen, W. L., 2004. The Many Roles of Computation
in Drug Discovery. Science 303, 5665-5682.
Karp P., Lee T., Wagner V., 2008. BioWarehouse:
Relational Integration of Eleven Bioinformatics
Databases and Formats. In Data Integration in the
Life Sciences, LNCS 5109, Springer Berlin /
Heidelberg.
Kirchmair, J., Distinto, S., Schuster, D., Spitzer, G.,
Langer, T. and Wolber, G. (2008) Enhancing drug
discovery through in silico screening: strategies to
increase true positives retrieval rates, Current
medicinal chemistry, 15, 2040-2053.
Köppen, H., 2009. Virtual screening - What does it give
us? Curr Opin Drug Discov Devel., 12(3), 397-407.
Krovat, E.M., Steindl T., Langer, T., 2005. Recent
Advances in Docking and Scoring, Current Computer
- Aided Drug Design, 1, 93-102.
Lala, D.S. 2005. The liver X receptors. Curr Opin Investig
Drugs 6, 934-43.
Maron, O., T. Lozano-Perez, T., 1998. A framework for
multiple-instance learning. In Advances in Neural
Information Processing Systems (NIPS), pages 570–
576. MIT Press.
Page, D., Craven, M., 2003. Biological applications of
multi-relational data mining. SIGKDD Explorations
5(1): 69--79.
Spencer, T.A. et al. 2001. Pharmacophore analysis of the
nuclear oxysterol receptor LXRalpha. J Med Chem 44,
886-97.
Winkler D.A., 2002. The role of quantitative structure-
activity relationships in molecular discovery. Briefings
in Bioinformatics 3, 73-86
Witten, I., and Frank, E., 2005. Data Mining: Practical
Machine Learning Tools and Techniques (Second
Edition), Morgan Kaufmann.
A KDD APPROACH FOR DESIGNING FILTERING STRATEGIES TO IMPROVE VIRTUAL SCREENING
151
