
Threats and Opportunities for the Clinical Investigation of High-risk
Medical Devices in the Context of the New European Regulations
L. Pazart
1,2
, S. Pelayo
1,3
, T. Chevallier
1,4
, G. Gruionu
5,6
, P. Mabo
1,7
, Y. Bayon
8
, F. Barbot
1,9
,
T. Lihoreau
1,2
, C. Roussel
1,10
, N. Maglaveras
11
, E. Lekka
11
, H. A. Ferreira
12
, I. Rocha
13
, L. Geris
14
and C. Lavet
1,
*
1
Inserm, Tech4Health, FCRIN F-31000 Toulouse, France
2
Univ. Franche-Comté, Inserm CIC1431, CHU Besançon, F-25000 Besançon, France
3
Univ. Lille, Inserm, CHU Lille, ULR 2694, METRICS, F-59000 Lille, France
4
IDIL- Nimes University Hospital and INSERM IDESP, F-30000, Nimes, France
5
Krannert Institute of Cardiology, Indiana University School of Medicine, Indianapolis, IN 46202, U.S.A.
6
INCESA Institute, Faculty of Mechanics, University of Craiova, Craiova 200512, Romania
7
Univ. Rennes, CHU de Rennes, Rennes, France
8
Medtronic, Sofradim Production, Trévoux, France
9
CIC 1429 Inserm, Hôpital Raymond Poincaré APHP, Garches, France
10
CIC 1415, CHU Tours, Inserm, University of Tours, France
11
Lab of Medical Informatics, Aristotle University of Thessaloniki, Thessaloniki, Greece
12
Instituto de Biofísica e Engenharia Biomédica, Faculdade de Ciências da Universidade de Lisboa, Portugal
13
Lisbon School of Medicine & Cardiovascular Centre, Universidade de Lisboa, Lisboa, Portugal
14
Biomechanics Research Unit, GIGA In Silico Medicine, University of Liège, Belgium
Keywords: High-risk Medical Device, Medical Device Regulation, Clinical Investigation, EU MDR 2017/745,
Regulatory Science.
Abstract: This position paper analyses the threats from the current situation of the clinical investigation to the
expectations of the new European regulations focusing on high risk medical devices (HRMDs). We present
also some opportunities to improve the feasibility and quality of clinical investigation. In summary,
investigation protocols of medical devices, advised and authorized by the competent authorities, are few and
heterogenous. There is a lack of quality in the existing studies, a lack of methodological knowledge and
consequently high expectations for assistance from those involved in the design of clinical study protocols
on HRMD. Guidance that is specific to the different type of devices is missing. Adaptive designs, pragmatic
trial, usability methods, computer modeling and real world data are gaining more and more traction for
assessing the safety and performance of high risk medical devices from a regulatory view- point.
*
EVAL-HRMD Project Consortium
1 INTRODUCTION
A series of major scandals have recently eroded
public confidence in the way high-risk medical
devices (HRMDs) are evaluated and monitored. Of
course, these situations have led to the withdrawal of
products from the market and legal actions have
been taken to sanction not only unscrupulous
manufacturers but also the notified bodies who issue
the famous ‘CE marking’ required to introduce new
products on the European market. By the end of
2018, the International Consortium of Investigative
Journalists' ‘implant files’ investigation shed light on
the way manufacturers can obtain the right to market
medical devices in Europe. These situations
highlight the weaknesses and failings of the health
control system for launching and monitoring
HRMDs. And yet, both patients and physicians want
to ensure that knowledge on innovation can
guarantee safe and efficient use of the new product.
New European regulations on medical device
(EU Medical Device Regulation 2017/745) will
come into effect in the spring of 2021. These new
regulations set forth new, improved rules to
strengthen clinical evidence, particularly for
HRMDs for which clinical investigation is
compulsory.
274
Pazart, L., Pelayo, S., Chevallier, T., Gruionu, G., Mabo, P., Bayon, Y., Barbot, F., Lihoreau, T., Roussel, C., Maglaveras, N., Lekka, E., Ferreira, H., Rocha, I., Geris, L. and Lavet, C.
Threats and Opportunities for the Clinical Investigation of High-risk Medical Devices in the Context of the New European Regulations.
DOI: 10.5220/0010382902740284
In Proceedings of the 14th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2021) - Volume 1: BIODEVICES, pages 274-284
ISBN: 978-989-758-490-9
Copyright
c
2021 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

This regulatory landslide represents a big
challenge for European Health SMEs (some 25,000
companies, representing 95% of the MedTech sector
in Europe) to maintain their competitiveness and
capacity for innovation, with limited internal
resources; especially in clinical trials skills. The
impact of Medical Device Regulation (MDR)
entering into force and the economic crisis linked to
the Covid-19 on the sustainability of these
companies has not yet been analyzed.
Updating clinical evaluation strategy and reports
to meet the new European requirements will require
major efforts for most manufacturers selling on the
EU market. Given the wide range of medical devices
(MD) available on the market and their countless
variations in design features, treatment goals and
targeted patient groups, setting a single standard
study protocol seems unfeasible.
This paper analyses the threats arising from the
current situation to the expectations of the new
European regulations focusing on HRMDs. We
present also some opportunities to improve the
feasibility and quality of clinical investigation.
2 DEFINITION AND
SPECIFICITIES OF HIGH-RISK
MEDICAL DEVICE
Classification of Medical Device is risk based, that
is, the risk the device poses to the patient and/or the
user is a major factor in the class it is assigned.
Three classes are defined, from Class I including
devices with the lowest risk to Class III including
those with the greatest risk (EU Medical Device
Regulation 2017/745). Device classification depends
on the intended purpose of the device, but also upon
indications for use and targeted population. Class III
devices usually sustain or support life, are
implanted, or present potential unreasonable risk of
illness or injury. Examples of Class III devices
include implantable pacemakers and breast implants.
Around 10% of medical devices fall under this
category.
High-Risk Medical devices (HRMDs)
correspond to current class III and implantable
devices. Many tools based on the annex VIII of the
MDR assist in the risk classification (class I, IIa, IIb
or III) of the product. But high risk and class III are
not necessarily totally overlapping. Other devices
may be high-risk and a variety of factors can
participate in the definition of a high-risk medical
device, such as a specific anatomical location for its
use, the implantable nature of the device, the use of
innovative or untested technologies or materials. The
implementation of any device can also be high-risk,
due to:
the vulnerability of the patient himself (e.g.:
children, pregnancy, chronic disease, aged)
the difficulty and delicacy of handling
the operator's dexterity and experience
(including the patient himself, his relatives or
health care staff)
the material environment in which the act
linked to the device will be performed
the potential complications of the procedure
performed.
HRMDs have particularities that make the
conduct of clinical investigations difficult, such as
long-term use and unknown interactions with the
human body, the means of explanting and replacing
implantable devices, the human-machine interface,
the management of data-flow generated, etc.
Although these issues are taken into account in
usability standards (IEC 62366-1 and IEC 60601-1-
6) and many methods of Human Factor Engineering
(HFE) have existed for years, they seem to be
underused (BSI 2016).
3 THREATS ARISING FROM
THE CURRENT SITUATION IN
EUROPE
3.1 The Loss of Europe’s
Attractiveness for Carrying out
Clinical Studies
The complexity of the regulatory process for
HRMDs is partly due to a significant fragmentation
of the global market. Namely, many countries have
their own set of rules around the world (Heneghan,
2012). A new device classified into Class III in
Europe may very well be considered a Class II
device according to a 510k procedure without the
need for a clinical investigation in the United States,
which is easier for businesses.
From our analysis (Figure 1) of annual
declarations of interventional studies on the website
clinicaltrials.gov, we note an increase of 16%
worldwide for medical devices over the last five
years (versus 9% for drugs), while Europe is
experiencing stagnation in the number of studies on
medical devices (+ 2%) and a decrease in the
number of studies on drugs (-5%).
Threats and Opportunities for the Clinical Investigation of High-risk Medical Devices in the Context of the New European Regulations
275
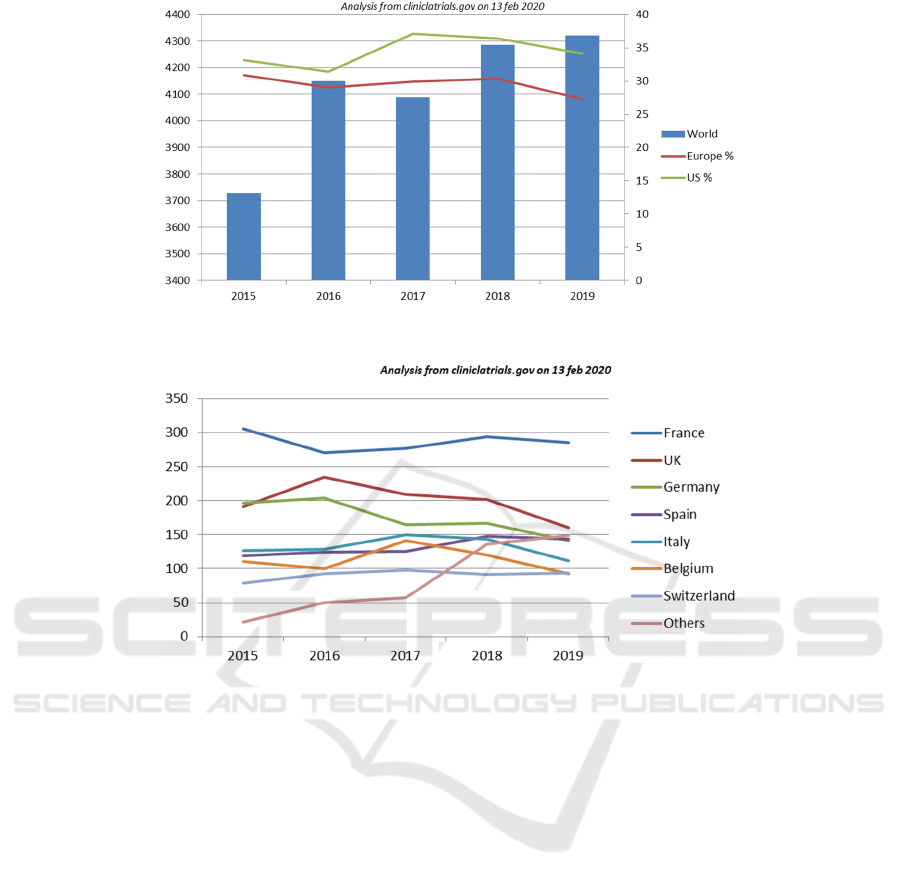
Figure 1: Evolution of clinical trials on medical devices worldwide and representative share for the USA and Europe (in %).
Figure 2: Distribution of Clinical trials on Medical Devices in Europe from 2015 to 2019.
Overall, one study on a medical device starts for
every 3 clinical trials starting on a drug. This has
been stable over the last five years worldwide
whereas, in Europe, this ratio which was identical to
the worldwide figure five years ago, is now
approaching a ratio of 1: 2 (3 medical device studies
for 8 drug studies in 2019).
The figure 2 shows how the initiation of
interventional clinical trials for medical devices has
slowed down in the different member states of
Europe (-10% in 2019 compared to the previous
year), and in particular from the year of publication
of the European regulation 2017/745, and the
dropout rate in international competition,
particularly in relation to the US.
3.2 The Current Observation on the
Lack of Quantity & Quality of
Clinical Studies on MD
Moreover, manufacturers have plenty of leeway and
different interpretations in performing or not the
clinical studies required to obtain their CE marking.
Most of them do the clinical evaluation of their
device based on data in the literature and
assimilation with predicates already on the market.
In 2017 the French health authority (ANSM)
registered CE markings for more than 15,293 new
medical devices (44% class II or III CE marks)
while, at the same time, this same authority only
issued 93 authorizations for new clinical trials of
medical devices.
The Iqwig (Independent German Institute for
Quality and Efficiency in Health Care) assessed the
methodological quality of 122 medical device
evaluation study projects submitted to the Berlin
ethics committee from March 2010 to December
2013 (Sauerland 2019). Of these 122 studies, 69%
were planned before marketing and 57% were
randomized controlled trials (RCTs). While only
half of the studies sought to demonstrate the
effectiveness of the medical device, in the other
studies the main objective stated was safety (18%),
performance (12%), patient-related benefits,
feasibility or user satisfaction.
ClinMed 2021 - Special Session on Dealing with the Change in European Regulations for Medical Devices
276
A European study by Olberg et al. highlights the
low level of evidence and the poor quality of studies
in the files submitted for the registration of HRMDs
with European technology agencies over the 2010-
2015 period (Olberg 2017). Their results concluded
that only 9% of these files had a very high level of
clinical evidence (meta-analysis but most of them
had pooled effect sizes driven up by a few
randomized control trials (RCT) of low-to-moderate
quality) and only 29% had a high level of evidence
(RCT). Overall, 61% of clinical studies had a
moderate to low level of evidence.
3.3 Imprecise Recommendations for
Conducting Adequate Clinical
Studies
The European Commission provides a range of
guidance documents to assist stakeholders in
implementing the regulations related to medical
devices (MEDDEVs guides). These guides promote
a common approach to be followed by the
manufacturers and Notified Bodies involved in
conformance assessment procedures. Revision 4
(MEDDEV 2.7/1 rev.4, June 2016) is more
prescriptive and requires manufacturers to provide
greater quantity and quality of information for
clinical evaluations. The first set of guidelines
(MEDDEVs guides) was recently updated and
clarified by the European Commission’s Medical
Device Coordination Group (MDCG 2020). The
MDCG posted new guidance (during the year 2020)
on clinical evaluation and evidence for devices and
postmarket clinical follow-up plans, representing for
us the basis to be completed by future guidelines
dedicated to HRMD:
However, these MDCG guidelines give general
advice, and miss the operational details needed to
adapt the design of studies and statistical analyses to
the characteristics of innovative technologies. The
most appropriate, least burdensome paths for
gathering clinical data to support marketing approval
for HRMDs are as varied as the devices themselves;
so more operational guidance are needed.
With the exception of a handful of cardiology
devices, available guidelines (from EU directives,
MDCG newly edited guides or national
transcriptions) and ISO/FDIS 14155 remain mostly
vague and imprecise in describing how to conduct a
clinical investigation and consider the clinical
evidence. Instead, the guidelines let manufacturers
choose how to create their clinical study protocols.
The manufacturers in charge of evaluating their
devices are asked to improve the process without
having the keys or knowledge to do so.
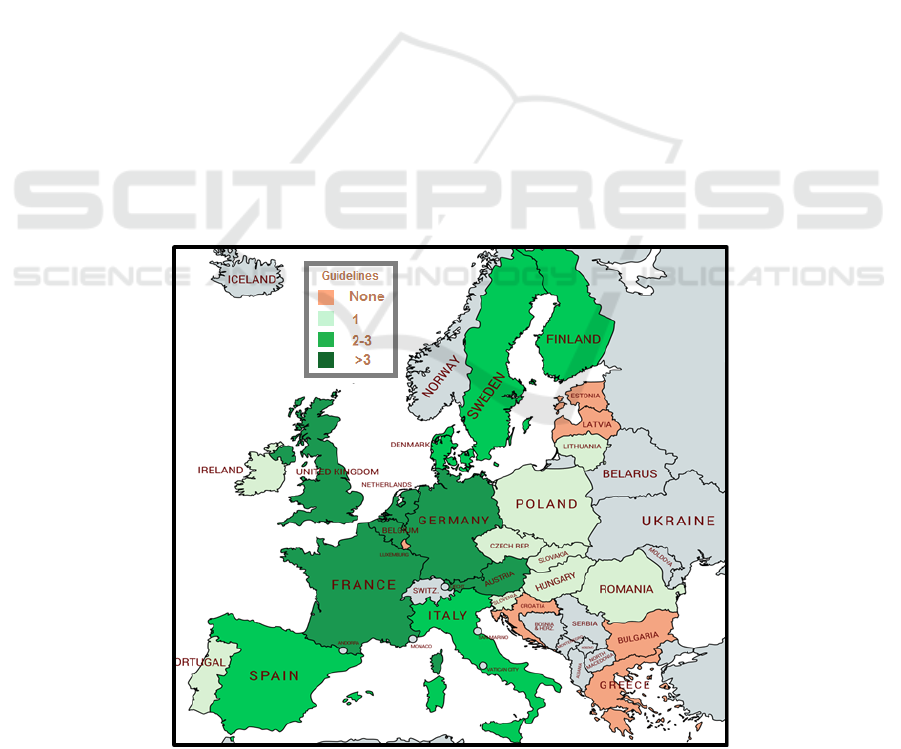
Figure 3: Number of guidelines on clinical evaluation of medical device per member state (Brunotte 2020).
Threats and Opportunities for the Clinical Investigation of High-risk Medical Devices in the Context of the New European Regulations
277

3.4 Heterogeneity of Advice in the
European Member States
In addition, broadly speaking, the MDRs leave the
organization of clinical investigation protocol
assessment and applicable authorization procedures
to the discretion of the Member States. Different
guidelines have been developed by member states
(figure 3, from Brunotte, 2020) but standard
methodologies are lacking. Those who should be
doing clinical studies on HRMDs -first of all,
manufacturers- have little visibility on what should
be done as well as lacking the required resources and
time.
Notified bodies must then request expert advice
to scrutinize manufacturers’ clinical evaluation
assessment report on HRMDs. Expert opinion does
not ensure a high level of clinical evidence and does
not guarantee a high level of reproducibility.
3.5 Regulation Is Uniform Disregards
Technological Characteristics and
the Evolution of the Devices
The same clinical evaluation requirements apply to
an English stick, a wheel chair, a hip prosthesis or a
connected pacemaker. Passive prostheses and active
implantable medical devices cover a large range of
medical applications and patient needs. These two
groups of HRMD exemplify very different R&D
situations.
R&D of passive prostheses, for example, mainly
involves the study of cells, their components,
complex tissues and organs and their interactions
with natural and synthetic materials. R&D of
implanted passive prosthetic devices also involves
developing and characterizing the materials used to
measure, restore, and improve physiological
functioning. These devices include coated stents,
bio-valves, joint replacements and cellular bone
grafts.
A second group is composed of active
implantable medical devices (AIMDs), (European
directive 90/385/EEC), mostly manufactured by
large international companies with considerable
technological resources (such as St Jude, Medtronic
and Becton Dickinson). AIMDs cover many
different clinical applications such as implantable
defibrillators, neuromuscular stimulators,
neuromodulators, cochlear implants and gastro-
intestinal pacemakers. One of the best-known
AIMDs is the cardiac pacemaker, introduced over 40
years ago, to deliver a controlled, rhythmic electrical
stimulus to cardiac tissue. AIMDs have shown an
impressive evolution over the last 20 years, not only
in size and weight (which has been reduced by a
factor of 10) but also in reliability, power
consumption and physiological functionality.
Specific recommendations have been issued from
learned societies of cardiology.
New advances in this type of devices are
expected with embedded algorithms of increasing
complexity, including adaptive stimulation
scenarios, diagnostic functions, data collection and
transmission, as well as remote multiprogramming
through a wireless link. Telemedicine may then
facilitate diagnosis and care over distances and
remote patient monitoring may lead to better home
care e.g. a pacemaker implanted in a patient, the
patient goes home, and the doctor monitors from a
distance. Patients may also access health information
via web portals, accessible anytime, anywhere.
Consequently, the traditional healthcare model of
patients traveling to see their doctor and being
diagnosed and treated inside hospital walls is no
longer the only relevant model.
The MDR does not differentiate between these
different types of devices, despite their different
characteristics and history of development
3.6 The Lack of Consideration of the
Characteristics of HRMDs for
Their Clinical Evaluation
Similar to drug evaluation, regulators around the
world generally prefer evidence from RCTs when
deciding whether to authorize the marketing of new
medical devices. However, RCTs are time
consuming and require significant financial
resources which are often underestimated; this is
particularly dangerous for SMEs with fragile
economic statuses Above all, this type of trials of
medical devices are difficult to perform for a
number of reasons:
a device implementation is a complex
intervention and the outcomes of the
intervention are generated by the combination
of varied factors involved – e.g. the device, the
clinicians implementing it, the training, the
clinical condition of the patient receiving it, …
the absence of comparators available on the
market
the device often evolves during the clinical trial
due to direct feedback from first end-users,
the difficulty of randomization due to the small
sample size of the target population,
ClinMed 2021 - Special Session on Dealing with the Change in European Regulations for Medical Devices
278

the operator (surgeon, cardiologist, radiologist
…) cannot be blinded to the type of device
implanted, and no placebo exists except for
devices that can be switched on and off
remotely, but “sham operations” are almost
never ethical because the patient experiences
the risk of the intervention but no benefit.
Overall, clinical studies on medical devices are
therefore far from systematic to run, and when
carried out, they do not present the level of
excellence (RCTs) expected by regulators to judge
clinical evidence. But is this requirement for level of
evidence really justified for all types of medical
devices?
3.7 New Digital Health Developments
Have Not yet Been Anticipated in
MDR
Nowadays, m-Health (mobile health) apps are
further widening the scope of how health services
can be delivered and, more importantly, these
technological advances are challenging traditional
healthcare services. The vision that emerges from
this is a health continuum (from healthy individuals
to seriously ill patients), and to cope in the
continuum of our lives we need Connected Health.
The Connected Health paradigm covers that
continuum and includes healthy individuals, those at
risk and chronically ill patients. In Connected
Health, individuals are equal partners with the
healthcare professionals and take part in managing
their own health.
p-Health (personalized health) and m-Health
represent these new domains of application for
information technology in the field of healthcare.
Over the next decade it will be a huge challenge to
propose new services to citizens and the right
regulation. These technologies produce data from
real-world settings. In addition to regulations, in
particular with the General Data Protection
Regulation (GDPR), the production and analysis
with AI of big data is actively studied in order to
develop new knowledge that has so far been
unaffordable.
3.8 High-risk Concept Poorly
Conceived in the MDR
Only three quotations of the word "high-risk" appear
in the 175 pages of the MDR, without any definition.
The clinical evaluation and assessment of level
of risk should incorporate all risk factors in the use
of HRMD and human factor issues.
4 OPPORTUNITIES FOR
CLINICAL INVESTIGATION
DESIGN
The European Commission has launched many calls
for projects on computer modeling, usability
methods, real world data processing and innovative
medical devices. Some of these will be useful for the
future.
Progress in computer modeling and simulation
applied to disease management is a European
strength and various Decision Support Systems have
been developed for different medical disciplines.
Through its new initiatives on digital health and care
within the Digital Single Market policy, the
European Commission aims to leverage the potential
of big data and high-performance computing for the
emergence of new personalized prevention methods
and treatments.
The economic aspects will be addressed in
existing European initiatives on the subject (e.g.
TBMED, MedTechHTA, Impact-HTA projects).
4.1 New Methodological Pathway for
Clinical Studies on HRMDs
The most important factor for successful marketing
approval, practitioner adoption, and safe use of
higher-risk medical devices is robust clinical
evidence.
In the United States, computer modeling and
simulation (i.e., in silico methods) are gaining more
and more recognition from regulatory boards for the
evaluation of the safety and performance of medical
devices. For example, from 2002 to 2019 in the
USA, at least 21% of the 565 pre-market approval
(PMA) applications for HRMDs had computational
modeling efforts provided in the Summary of Safety
and Effectiveness Data (Morrison 2018, 2019). For
the past few years, the FDA has been accepting
regulatory files including digital models, adaptive
studies, hybrid trials, real-world data and experience.
The FDA has thus developed numerous general
guides on these subjects to help medical device
manufacturers carry out adequate studies to obtain
their product launches (FDA 2010, 2018, 2020).
Given the shortcomings of the MEDDEV and
MDCG guidelines and of the ISO/FDIS 14155
Threats and Opportunities for the Clinical Investigation of High-risk Medical Devices in the Context of the New European Regulations
279

standards in terms of the expected clinical study
design, further issues concern the following
considerations:
alternatives to classical randomized controlled
trials, including pragmatic trials and adaptive
design
alternatives to frequentist approaches
integration of Human Factors and usability
study methods
the place of computer modelling and
simulation models (in silico models and trials)
the use of real-world data with new analytical
capabilities and mathematical models.
a deal with companies to get real world data
(RWD) generated by HRMD against freely use
of academic simulation models
4.2 Adaptive Methodologies and
Pragmatic Trials Have Been
Developed as an Alternative to the
Classical RCT Design
Even though the legislation, particularly American
legislation with the Food and Drug Administration
(FDA), qualifies adaptive methodologies as
“modern” and “new” methods, a large number of
these concepts are old but have remained unused for
many years especially by the European notified
bodies for MDs evaluation.
Methodologists propose using tracker design
trials (Lilford 2000), sequential trials (Hamilton
2012), ‘Multi-Arm Multi-Stage’ trials (Wason 2014,
Wathen 2017),
pragmatic trial (Ford 2016, Loudon
2015, Thorpe 2009, Simon 2019, Gamerman 2019)
and adaptive trials (Simon 2013, Meurer 2016,
Magirr 2016, Lai 2019) to take technological
evolution into account and accelerate clinical
development and product launching whilst allowing
early terminations (futility/efficacy) or protocol
adjustments (evolution/suppression of an arm).
These trials rely on planned interim analyses which
allow the investigator to glean useful information for
adapting the strategy. They are particularly relevant
to the context of HRMD clinical evaluation.
With adaptive methods it is also possible to
strengthen the clinical evaluation of medical devices
by authorizing the analysis of multiple evaluation
criteria, carrying out several intermediate analyses,
early terminations in the event of inefficacity,
allocating patients to the most promising arms, re-
evaluating the sample-size and, more especially,
redefining the target population. With these methods
it is also possible to combine the early exploratory
phases with the demonstrative phases which may
help to accelerate and optimize the development and
implementation of innovative devices. When certain
centers only use one of the two techniques under
study and do not know the other technique, or only
master one technique and the result is operator-
dependent, it is possible to use a trial based on
expertise or a cluster trial (or a Stepped Wedge
Cluster trial, Barker 2016) to increase the
participation of doctors and the reliability of the
evaluation. When one arm in the study is less
attractive than the other, studies may be carried out
according to a Zelen plan (Zelen 1990) or according
to a complete cohort pattern. These types of trials
introduce flexibility in the attribution of treatments
and allow better acceptability of the randomization
by the patients and also give us the possibility of
adjusting the results to the randomization. Group
sequential design and adaptive sample-size
adjustment are often used to make study durations
shorter and include a smaller number of subjects.
Nevertheless, there has been criticism of these
adaptive designs and it will be important to analyze
the biases and added value of these proposals, their
acceptability by the stakeholders and their
admissibility by the European authorities.
For the past few years, the FDA has been
accepting regulatory files including digital models,
adaptive studies, hybrid trials design, real-world data
and experience (Guetterman 2017, Campbell 2019).
FDA has thus developed numerous guides on these
subjects to help manufacturers of medical devices to
carry out adequate studies to obtain a marketing of
their products (FDA 2010, 2018, 2020).
4.3 Bayesian Approaches May be used
to Implement and Analyze Clinical
Trials
Bayesian approaches give the possibility of
combining prior information before the trial
(previous studies, expert opinion, literature…) and
current information during the trial to formulate or
reformulate decision-making rules (Campbell 2011,
Ribouleau 2011, Wei 2018).
In a Bayesian clinical trial, any uncertainty about
a parameter is described according to probabilities,
which are then updated during data-collection for the
trial. The probabilities are set beforehand based on
previous data and the probabilities are estimated a
posteriori from the data obtained during the trial
(Pennello 2008). There are no statistical tests but the
probability of the treatment under experimentation
being effective has a 95% credibility threshold.
However, it is very important that the a priori
ClinMed 2021 - Special Session on Dealing with the Change in European Regulations for Medical Devices
280

information used does not over influence the final
result (sensitivity analysis required). The quality of
information supplied a priori is therefore a key
element in the credibility of results.
4.4 Human Factors Engineering
There are a variety of human factors and usability
evaluation methods (Genise, 2002) for all stages of
design and development, from product definition to
final design modifications like cognitive modeling
methods, inspection methods, inquiry methods,
prototyping methods, usability testing etc. Certain
methods use data from users, while others rely on
usability experts. When choosing a method, cost,
duration and appropriateness should be considered.
4.5 In Silico Modelling
The beginning of the 21st century saw the birth of a
completely new way to investigate living organisms
through computer simulations, called in silico
medicine. Over the last 15 years, significant efforts
have been made to build numerical patient models
from multimodal images, for instance, for surgical
planning and image-guided surgery.
Initially released in 2007, the Virtual Family
(https://www.fda.gov/about-fda/cdrh-offices/virtual-
family) is a set of four highly detailed, anatomically
correct whole-body models of an adult male, an
adult female, and two children. The Virtual Family
project was carried out in collaboration between the
FDA and academic or private European partners
from Erlangen, Germany, and Zürich, Switzerland.
Currently, the Virtual Family models are used for
electromagnetic, thermal, acoustic, and
computational fluid dynamics simulations. Examples
of applications of electromagnetic and thermal
simulations are the assessment of the safety of active
and passive medical implants in a Magnetic
Resonance Imaging (MRI) environment and the
evaluation of the safety and efficacy of ablation
devices. Since the end of 2014, the Virtual Family
has been regularly used in medical device
submissions to the FDA.
The Virtual Physiological Human (VPH) is an
initiative developed over the last decade and
supported by the European Commission to create a
computational framework designed to facilitate the
understanding of the integrative function of
molecules, cells, tissues, and organs and, by this, to
construct a multiscale in silico model of the human
physiology (Viceconti 2008). The collective
framework will make it possible to share resources
and observations formed by institutions and
organizations, creating disparate but integrated
computer models of the mechanical, physical and
biochemical functions of a living human body. VPH
is a framework which aims to be descriptive,
integrative and predictive. The framework consists
of large collections of anatomical, physiological, and
pathological data stored in digital format, with
predictive simulations developed from these
collections and services intended to support
researchers in the creation and maintenance of these
models, and also the creation of end-user
technologies to be used in clinical practice.
The validation of in silico clinical trial models
poses relevant theoretical problems. However, these
have been discussed in specialized publications
(Coveney, 2014) and a standardization committee
(ASME V&V-40 verification and validation in
computational modelling of medical devices), which
worked on some codified guidelines (Popelar, 2013).
A key aspect, which was promoted within the
Medical Device Innovation Consortium (Kampfrath
2013), but that emerged again and again during the
Avicenna consensus process, is that of model
credibility. The process to ensure that a predictive
model is indeed accurate in its predictions is
somehow at the center of a paradox. Models are
usually developed to predict things that cannot be
easily measured, so how do we know how accurate
these predictions are?
4.6 Use of Real-World Data
The use of computers, mobile devices, wearables
and other biosensors gathering huge amount of
health data has been rapidly accelerating. These data
hold potential to allow us to better design and
conduct clinical trials and studies in the healthcare
setting to answer questions previously thought
infeasible. In addition, with the development of
sophisticated, new analytical capabilities, we are
able to better analyze these data (Kumsa 2018,
Sherman 2016).
The increasing availability of data generated by
such devices poses challenges regarding
management and data workflows. The use of
artificial intelligence algorithms in medical devices,
can lead to undetermined risks for users, and require
a proper framework for development and validation.
Progress, particularly in computing and AI, data and
wearable accessibility, is often made at a much
faster rate than guidelines and recommendations are
issued.
Threats and Opportunities for the Clinical Investigation of High-risk Medical Devices in the Context of the New European Regulations
281

5 CONCLUSIONS
In summary, investigation protocols of high risks
medical devices, advised and authorized by the
National competent authorities in Europe, are few
and heterogenous. There is a lack of quality in the
existing studies, a lack of methodological knowledge
and consequently high expectations for assistance
from those involved in the design of clinical study
protocols on HRMD. Guidance that is specific to the
different type of devices is missing.
The “new” EU MDR 2017/745 coming into
effect in May 2021, by obliging clinical
investigation and post-market follow-up, offers the
opportunity to develop novel pathways for the
clinical development of HRMDs. In particular, there
is a perceived lack of knowledge and training in
clinical trial skills in European medical device
companies who could greatly benefit from a
clarification of expectations linked to the MDR for
HRMDs..
Nevertheless, the available guidelines (from EU
directives, MDCG new guides or national
transcriptions) remain vague and imprecise, also
many companies are leaving Europe due to the
complexity and imprecision of MDR. They move to
the US where the regulatory pathway is clearer &
faster.
The risks of this situation in Europe are
reinforced by many threats:
the current observation on the lack of
quality clinical studies,
the specificities of HRMD by medical
speciality,
the economic fragility of European HRMD
companies (95% SMEs),
the high expectations of safety on the part
of patients and healthcare professionals,
the loss of attractiveness of Europe for
carrying out clinical studies,
the desire for a "smooth transition" from
directives to MDR without real means.,
the attribution of a CE marking by various
private structures,
the diversity of approach from one notified
body to another, and the absence of a
centralized procedure
At both European and national level, before and
after marketing, a new balance needs to be found
between the need for rigorous evidence and the real
world complexity of gathering such evidence; a
balance between strict regulation and high levels of
evidence for high risk medical devices, and the
possibility of other types of evidence for devices
associated to lower levels of risk.
Adaptive designs, pragmatic trial, usability
methods, computer modeling and real world data are
gaining more and more traction in the United States
for assessing the safety and performance of medical
devices from a regulatory view- point. The European
Commission has launched many calls for projects on
these subjects, generating new knowledge and new
teams of experts. Expectations from stakeholders of
clinical investigations tend to bring these experts and
knowledge together to prioritize the methods and
develop useful guidelines for those who wish to set
up clinical studies on HRMDs.
ACKNOWLEDGEMENTS
We would like to thank all those who contributed to
the preparation of the Eval-HRMD project, and
especially Joanna Baranowska, Marine Beaumont,
Regis Beuscart, Patrick Boisseau, Gaelle Brunotte,
Guy Carrault, Jacques Demotes, Marlene Durand,
Maria Dutarte, Jerôme Fabiano, Jacques Feblinger,
Joris Giai, Lucian Gruionu, Gemma Killeen, Amélie
Michon, Alexandre Moreau-Gaudry, Claire Nassiet,
Frédéric Patat, Pierre Pautre, David Orlikowski,
Delphine Smagghe, Dimitar Tcharaktchiev,
Chrystelle Vidal, Sebastian Wawrocki. The project
received a grant from the National Research Agency
to support the networking of partners.
REFERENCES
ASME - American Society of Mechanical Engineers -
standard V&V40 2018: “Assessing credibility of
computational models through verification and
validation: application to medical devices”
Barker, D, et al., 2016. Stepped wedge cluster randomised
trials: a review of the statistical methodology used and
available. BMC Med Res Methodol 6;16:6..
British Standards Institution (BSI), 2016 The growing role
of human factors and usability engineering for medical
devices, white paper.
Brunotte G., Beuscart, R., Pariset, A. Pazart, L. 2020 Place
of High-risk Medical Devices in European
Recommendations with a Focus on End-users. In
Proceedings of the 13th International Joint Conference
on Biomedical Engineering Systems and Technologies
- Vol 1: BIODEVICES, p 350-360
Campbell, G, 2011. Bayesian statistics in medical devices:
innovation sparked by the FDA. J Biopharm Stat.
21:871-87.
ClinMed 2021 - Special Session on Dealing with the Change in European Regulations for Medical Devices
282

Campbell B, Wilkinson J, Marlow M, et al. 2019
Generating evidence for new high-risk medical
devices. BMJ Surg Interv Health Technologies;
1:e000022
Coveney, P., Diaz, V., Hunter, P., Viceconti, M., Eds.,
2014. Computational Biomedicine. Oxford, Oxford
University Press.
FDA - Guidance for the use of Bayesian Statistics in
Medical Device Clinical Trials; Guidance for industry
and FDA Staff on February 5, 2010.
FDA - Guidance on the use of “real-world evidence” —
healthcare information from atypical sources,
including electronic health records, billing databases,
and product and disease registries — to assess the
safety and effectiveness of drugs and devices. 2018
FDA - Adaptive Designs for Medical Device Clinical
Studies, Guidance for Industry and Food and Drug
Administration Staff, Document issued on July 27,
2016, new version 2020 under comments.
Ford I, Norrie J. 2016 Pragmatic Trials N Engl J Med
2016; 375:454-463 DOI: 10.1056/NEJMra1510059
Gamerman, V., Cai, T. & Elsäßer, (2019) A. Pragmatic
randomized clinical trials: best practices and statistical
guidance. Health Serv Outcomes Res Method 19, 23–
35. doi.org/10.1007/s10742-018-0192-5
Genise P. 2002 “Usability Evaluation: Methods and
Techniques: Version 2.0” August 28,. University of
Texas.
Guetterman, TC, et al. 2017. The life cycles of six multi-
center adaptive clinical trials focused on neurological
emergencies developed for the Advancing Regulatory
Science initiative of the National Institutes of Health
and US Food and Drug Administration: Case studies
from the Adaptive Designs Accelerating Promising
Treatments Into Trials Project. SAGE Open Med..
Guideline on clinical Trials in small populations;
Committee for medicinal products for human use on
27 july 2006.
Hamilton, C,et al., 2012. Sequential design for clinical
trials evaluating a prosthetic heart valve. Ann Thorac
Surg, 93(4): 1162-1166.
HAS, Methodological choices for the clinical development
of medical devices; HAS evaluation report dated
October 4th, 2013.
HAS, Methodological specificities of the clinical
evaluation of a connected medical device (CMD);
HAS report on the elaboration of the guide on the
specificities of clinical evaluation, in view of its access
to reimbursement dated January 29th, 2019.
Heneghan C, Thompson M. Rethinking medical device
regulation. Journal of the Royal Society of Medicine.
2012;105(5):186-188.
Kampfrath, T., Cotten, S. W., 2013. The new collaborative
path in medical device development: The medical
device innovation consortium. Clinical Biochemistry
46, 1320-1322
Kumsa , Doe, et al. (2018) "Public regulatory databases
as a source of insight for neuromodulation devices
stimulation parameters." Neuromodulation:
Technology at the Neural Interface 21. 2: 117- 125 .
Lai, TL, et al., 2019. Adaptive enrichment designs for
confirmatory trials. Stat Med 38 (4):613-624.
Lilford, R, et al., 2000. Trials and fast changing
technologies: the case for tracker studies. BMJ, 320
(7226): 43–46.
Loudon K, Treweek S, Sullivan F, Donnan P, Thorpe KE,
Zwarenstein M et al. 2015 The PRECIS-2 tool:
designing trials that are fit for purpose BMJ; 350
:h2147
Magirr, D, et al., 2016. Sample Size Reassessment and
Hypothesis Testing in Adaptive Survival Trials. PLoS
One. 11(2):e0146465.
Meurer, WJ, et al., 2016. Attitudes and opinions regarding
confirmatory adaptive clinical trials: a mixed methods
analysis from the Adaptive Designs Accelerating
Promising Trials into Treatments (ADAPT-IT)
project. Trials. 29;17:373.
MDCG 2019-9 Summary of safety and clinical
performance A guide for manufacturers and notified
bodies - August 2019
MDCG 2020-1 Guidance on Clinical Evaluation (MDR) /
Performance Evaluation (IVDR) of Medical Device
Software
MDCG 2020-5 Clinical Evaluation - Equivalence. A guide
for manufacturers and notified bodies
MDCG 2020-6 Regulation (EU) 2017/745: Clinical
evidence needed for medical devices previously CE
marked under Directives 93/42/EEC or 90/385/EEC.
A guide for manufacturers and notified bodies
MDCG 2020-7 Post-market clinical follow-up (PMCF)
Plan Template. A guide for manufacturers and notified
bodies
MDCG 2020-8 Post-market clinical follow-up (PMCF)
Evaluation Report Template. A guide for
manufacturers and notified bodies
MEDDEV 2.7/1 rev.4 Guidelines on medical devices,
clinical evaluation: A guide for manufacturers and
notified bodies under directives 93/42/EEC and
90/385/EEC European Commission June 2016.
Morrison, Tina (2019): LyoHUB: Advancing Regulatory
Science with Modeling and Simulation at the FDA.
figshare. Presentation. https://doi.org/10.6084/m9.fig
share.9910832.v1
Morrison, T. M., et al., (2018). Advancing regulatory
science with computational modeling for medical
devices at the FDA's Office of Science and
Engineering Laboratories. Frontiers in medicine, 5.
Olberg B, Fuchs S, Panteli D, Perleth M, Busse R. 2017
Scientific Evidence in Health Technology Assessment
Reports: An In-Depth Analysis of European
Assessments on High-Risk Medical Devices. Value
Health. Dec;20(10):1420-1426.
Pennello, G, et al., 2008. Experience with reviewing
Bayesian medical device trials. J Biopharm Stat 18
:81-115.
Popelar, C. F., 2013. Verification & Validation in
Computational Modeling of Medical Devices (V&V-
40): ASME subcommitee and guide. FDA/NIH/NSF
Workshop on Computer Models and Validation for
Medical Devices. Silver Spring, MD.
Threats and Opportunities for the Clinical Investigation of High-risk Medical Devices in the Context of the New European Regulations
283

Ribouleau, L, et al., 2011. Bayesian statistical method was
underused despite its advantages in the assessment of
implantable medical devices. J Clin Epidemiol.
64:270-9.
Sauerland S, Fujita-Rohwerder N, Zens Y, Molnar S.
Premarket evaluation of medical devices: a cross-
sectional analysis of clinical studies submitted to a
German ethics committee. BMJ Open. 2019
Feb;9(2):e027041. DOI: 10.1136/bmjopen-2018-
027041.
Sherman RE & al. (2016) Real-World Evidence — What
Is It and What Can It Tell Us? N Engl J Med 2016;
375:2293-2297 DOI: 10.1056/NEJMsb1609216
Simon, N, et al., 2013. Adaptive enrichment designs for
clinical trials. Biostatistics 14(4):613-25.
Simon, G.E., Shortreed, S.M., Rossom, R.C. et al. (2019)
Principles and procedures for data and safety
monitoring in pragmatic clinical trials. Trials 20, 690.
doi.org/10.1186/s13063-019-3869-3
Thorpe KE, Zwarenstein M, Oxman AD, et al. 2009 A
pragmatic-explanatory continuum indicator summary
(PRECIS): a tool to help trial designers. J Clin
Epidemiol.;62(5):464‐475.
doi:10.1016/j.jclinepi.2008.12.011
Viceconti M, Clapworthy G, Van Sint Jan S. 2008 The
Virtual Physiological Human - a European initiative
for in silico human modelling -. J Physiol Sci.
Dec;58(7):441-6.
Wason JM, et al., 2014. A comparison of Bayesian
adaptive randomization and multi-stage designs for
multi-arm clinical trials. Stat Med. 33 (13):2206-21.
Wathen JK et al., 2017. A simulation study of outcome
adaptive randomization in multi-arm clinical trials.
Clin Trials 14 (5):432-440.
Wei, B, et al., 2018. A Bayesian analysis of small n
sequential multiple assignment randomized trials
(snSMARTs). Stat Med 20 ; 37 (26) :3723-3732.
WHO, 2019. Medical devices URL (accessed 12.18.19).
http://www.who.int/medical_devices/en/
World Health Organization (2014) WHO handbook for
guideline development, 2nd ed. World Health
Organization. 167 pages https://apps.who.int/iris/
handle/10665/145714
Zelen, M, et al., 1990. Randomized consent designs for
clinical trials: an update. Stat Med 9 (6): 645-56
ClinMed 2021 - Special Session on Dealing with the Change in European Regulations for Medical Devices
284
