Machine Learning Studies of Non-coding RNAs based on Artificially
Constructed Training Data
Mirele C. S. F. Costa
1,† a
, Jo
˜
ao Victor A. Oliveira
2 b
, Waldeyr M. C. da Silva
1,3 c
,
Rituparno Sen
4 d
, J
¨
org Fallmann
4 e
, Peter F. Stadler
4,5,6,7 f
and Maria Em
´
ılia M. T. Walter
1,‡ g
1
University of Bras
´
ılia (UnB), Brazil
2
Federal Institute of Bras
´
ılia (IFB), Brazil
3
Federal Institute of Goi
´
as (IFG), Brazil
4
Bioinformatics Group, Department of Computer Science, Interdisciplinary Center for Bioinformatics,
University of Leipzig, Leipzig, Germany
5
German Centre for Integrative Biodiversity Research (iDiv) Halle-Jena-Leipzig, Leipzig, Germany
6
Max Planck Institute for Mathematics in the Sciences, Leipzig, Germany
7
Santa Fe Institute, Santa Fe, U.S.A.
Keywords:
Small Nucleolar RNAs (snoRNAs), Non-Coding RNA Inference, Machine Learning, Chordate Genome.
Abstract:
Machine learning (ML) methods are often used to identify members of non-coding RNA classes such as
microRNAs or snoRNAs. However, ML methods have not been successfully used for homology search tasks.
A systematic evaluation of ML in homology search requires large, controlled, and known ground truth test
sets, and thus, methods to construct large realistic artificial data sets. Here we describe a method for producing
sets of arbitrarily large and diverse snoRNA sequences based on artificial evolution. These are then used to
evaluate supervised ML methods (Support Vector Machine, Artificial Neural Network, and Random Forest) for
snoRNA detection in a chordate genome. Our results indicate that ML approaches can indeed be competitive
also for homology search.
1 INTRODUCTION
Many distinct classes of non-coding RNAs (ncRNAs)
are known, each with specific function, in turn de-
pending on their spatial structure, sequence compo-
sition and length. In this contribution we focus on
small nucleolar RNAs (snoRNAs). They form a large
class of RNAs with lengths varying from 60 to 300
nucleotides that comprises two functionally and struc-
turally distinct subclasses, the H/ACA box and C/D
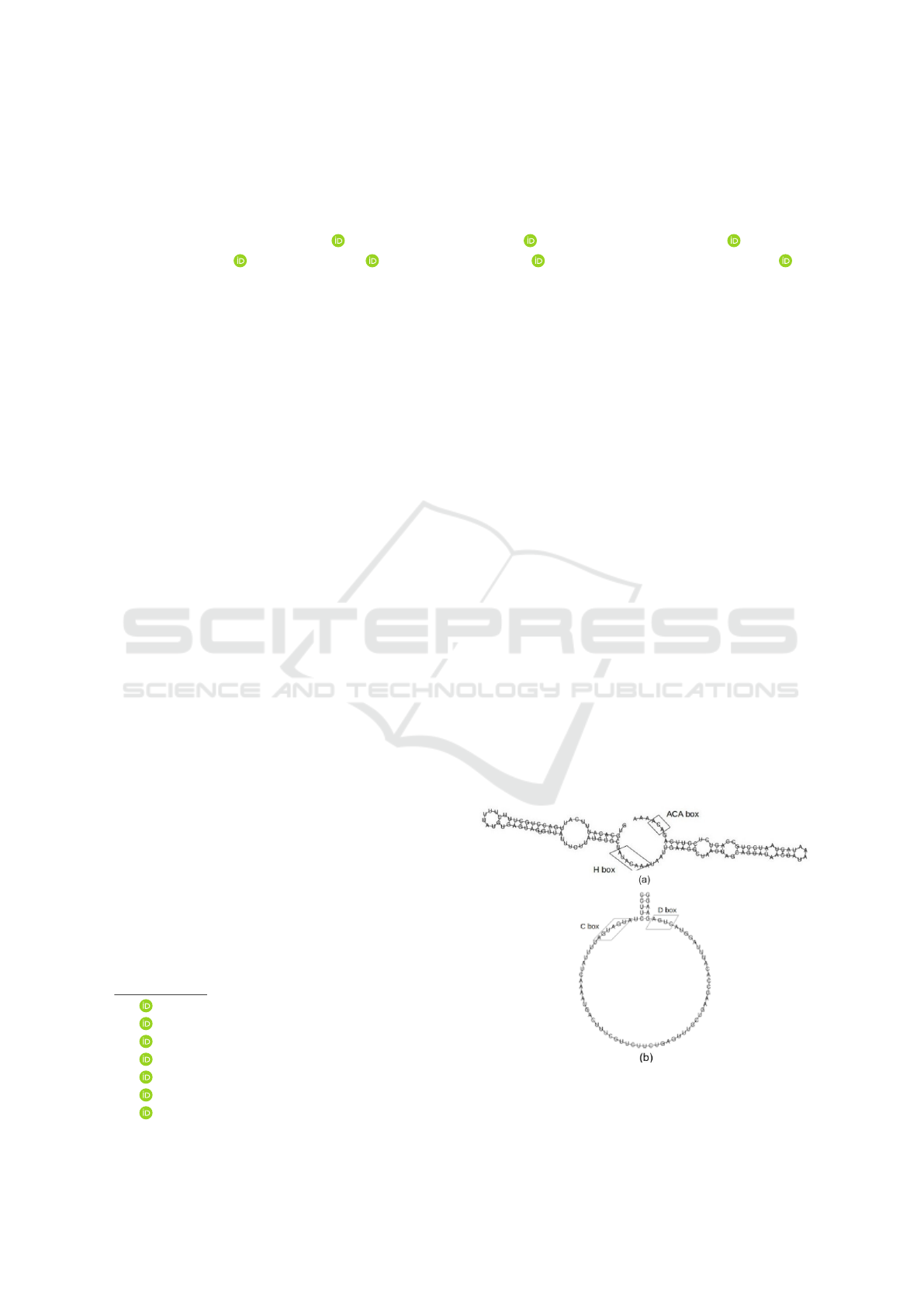
box (Falaleeva and Stamm, 2013) (see Figure 1). In
animals, snoRNAs are processed from introns of both
coding and non-coding host RNAs (Bratkovi
ˇ
c et al.,
2020).
a
https://orcid.org/0000-0002-1337-4672
b
https://orcid.org/0000-0003-3622-1334
c
https://orcid.org/0000-0002-8660-6331
d
https://orcid.org/0000-0001-5980-5565
e
https://orcid.org/0000-0002-4573-9939
f
https://orcid.org/0000-0002-5016-5191
g
https://orcid.org/0000-0001-6822-931X
There are two fundamentally different methods
to identify ncRNAs in genomic data: (1) Homol-
ogy search utilizes sequence similarity to a specific
Figure 1: Two-Dimensional structure of (a) H/ACA box
snoRNA; and (b) C/D box snoRNA (de Araujo Oliveira
et al., 2016).
176
Costa, M., Oliveira, J., C. da Silva, W., Sen, R., Fallmann, J., Stadler, P. and Walter, M.
Machine Learning Studies of Non-coding RNAs based on Artificially Constructed Training Data.
DOI: 10.5220/0010346001760183
In Proceedings of the 14th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2021) - Volume 3: BIOINFORMATICS, pages 176-183
ISBN: 978-989-758-490-9
Copyright
c
2021 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

query sequence. Since structure dictates function of
many RNAs, their spatial structures are often bet-
ter conserved than their sequences. Thus, homol-
ogy search methods for ncRNAs usually also take
structural similarity into account. Tools such as
infernal (Nawrocki and Eddy, 2013) indeed achieve
substantial improvements compared to sequence-only
methods such as Blast (Altschul et al., 1990), see
e.g. (Bartschat et al., 2014). However, by construc-
tion, only homologs of known ncRNAs can be found.
(2) Some ncRNAs, including transferRNAs, microR-
NAs, and snoRNAs, belong to larger families that
share both function and biogenesis and are conse-
quently recognizable by a set of characteristic se-
quence and structure features. The identification of
members of an RNA class is a classification prob-
lem that is typically solved by machine learning meth-
ods (Zhang et al., 2017; Barber, 2012; Zhang and Ra-
japakse, 2009). SnoReport 2.0 (de Araujo Oliveira
et al., 2016) implements such a classifier for snoR-
NAs. It extracts a combination of sequence and sec-
ondary features, the latter predicted by thermody-
namic folding (Lorenz et al., 2011), from a query se-
quence and employs a support vector machine (SVM)
for classification into the two main classes of snoR-
NAs: the H/ACA box and the C/D box. SnoReport
2.0 can be used to scan large DNA sequences, using
characteristic sequence and structure motifs to iden-
tify candidates that are passed to the SVM classi-
fier. Similarly, classifiers for miRNAs start from a
predicted precursor hairpin. This makes the classi-
fication problem fairly simple since the task merely
distinguishes whether the input sequence is exactly a
class member.
A closely related, but apparently much more diffi-
cult machine learning problem is to ask whether or not
a given sequence of fixed length contains a ncRNA of
a given class. An efficient solution to this version of
the ncRNA classification problem would provide an
alternative to homology search for large evolutionary
distance, where sequence similarity comes close to or
even falls below the detection limit. Anecdotal reports
on attempts to use machine learning for this task, e.g.,
(Waldl et al., 2018), however, have been discourag-
ing – albeit this may be a consequence of very small
training sets. A more systematic investigation into
the feasibility of machine learning as an alternative
to direct sequence comparison requires training and
test sets that are large and diverse enough. Further-
more, they have to cover a wide range of evolutionary
distances from closely related sequences to homologs
that have diverged beyond the detection limit for se-
quence alignment methods.
In this contribution, we present a method to gen-
erate in principle an arbitrarily large and diverse data
set of artificial ncRNAs, using snoRNAs as an exam-
ple. The key idea is to simulate the evolution of the
ncRNAs along a real or randomly generated phyloge-
netic tree, using a classifier for the ncRNAs of inter-
ests, here snoReport 2.0, to model selection. That
is, mutations are only accepted if they pass the clas-
sifier. The procedure thus “breeds” snoRNAs with
increasingly divergent sequences that are still recog-
nizable as snoRNAs. The artificial ncRNAs can then
be inserted into background genomes to produce re-
alistic data with perfectly-known ground truth to train
and benchmark homology search methods. Here we
consider Support Vector Machines (SVMs), Random
Forests (RFs), and artificial neural networks (ANNs)
as classification methods.
2 METHODS
We start in Section Initial Data with a description of
the biological data that we use as starting point and for
evaluation purposes. We then describe construction
of an artificial snoRNA data set (”Breeding” Artifi-
cial snoRNAs). The third part of this section (Feature
Extraction) summarizes the features used to evaluate
de ML methods. Finally, we provide a detailed evalu-
ation of ML approaches versus direct sequence com-
parison. On the top level, the workflow starting with
the initially acquired data can be subdivided into six
stages, which are also presented in figure 2:
1. Run snoReport 2.0 over the selected intron se-
quences to identify snoRNAs and their corre-
sponding C/D or H/ACA boxes.
2. Choose randomly representative snoRNA se-
quences from snoReport 2.0 output to apply
mutations;
3. Build mutation tree using the snoReport 2.0
output sequences composing sets with cumulative
percentage of mutations;
4. Extract features for each sequence, considering
the positive and negative sets obtained from the
mutation tree;
5. Construct datasets for ML algorithms with the
same number of instances for the positive (1) and
negative (0) sets.
6. Execute ML algorithms and analyze their results.
2.1 Initial Data
The source data used in the experiments was extracted
from the marine species Ciona intestinalis, obtained
Machine Learning Studies of Non-coding RNAs based on Artificially Constructed Training Data
177

Figure 2: Summary of the steps to build the dataset and
analyze the machine learning methods.
from the National Center for Biotechnology Informa-
tion (NCBI). We retrieved the genome and transcript
sequences in fasta format, and the genome annota-
tion in a GFF3 format. C. intestinalis is an attrac-
tive model for studying chordate origins and evolu-
tion since it has a compact genome, which is advanta-
geous for developmental evolutionary studies (Satoh,
2003). We used Blastn (Altschul et al., 1990) to lo-
cate the known snoRNA sequences from C. intesti-
nalis in the annotated intronic sequences, which were
retrieved using GFF-Ex (Rastogi and Gupta, 2014). A
cut-off of 80% sequence identity was employed. This
resulted in one H/ACA box snoRNA and two C/D box
snoRNAs, which served as starting points for our sim-
ulations.
Algorithm 1: Construction of the mutation tree.
Data: s// a snoRNA sequence intron// the
intron containing the snoRNA
sequence
Result: T // a mutation tree
1 mutate s: sMutated
2 replace s by a sMutated into the intron
3 if intron has a snoRNA then
4 if sMutated is identified as snoRNA in the same
locus of the original one then
5 sMutated is inserted into T , to be
successively mutated;
6 sMutated is stored in the positive set
corresponding to the tree level;
7 else
8 sMutated is not inserted into T and is stored
in a separate set;
9 sMutated is not inserted into T and is stored in
the negative set;
2.2 ”Breeding” Artificial snoRNAs
SnoReport 2.0 was used to identify snoRNA se-
quences and their corresponding C/D box or H/ACA
box classes in the carefully selected intron sequences
as described in subsection 2.1 (Fig 2-Step 1). From
those sequences, two representatives were chosen to
generate the mutation trees, one C/D box snoRNA
and one H/ACA box snoRNA (Fig 2-Step 2). The
total length of the intron and of the original snoRNA
are 833 and 96, for C/D box, and 173 and 1,577, for
H/ACA box, respectively. Algorithm 1 describes the
construction of the mutation tree and thus of the artifi-
cial snoRNAs. These are then used to define positive
and negative sets for the classification task (Fig 2-Step
3).
The root of the tree corresponds to one of the
representative snoRNA sequences s described above.
In each step, mutations (substitutions, deletions, and
insertions) are applied to s. The resulting mutated
sequence sMutated is then re-inserted into the in-
tronic sequences. We define sMutated as a synthetic
snoRNA, i.e., as a true positive, if it is recognized in
this context as a snoRNA by snoReport 2.0. Muta-
tion trees are constructed independently for each ini-
tial snoRNA. In each level of the tree we train at most
N sequences. The mutation process mimics a popu-
lation of fixed size with N = 3000 sequences for C/D
box and N = 2000 for H/ACA box to limit the compu-
tational resources. In order to obtain balanced trees,
the sequence to be mutated is chosen at random from
this population. The nodes of mutation tree are gener-
ated with 10 children. The positive set consists of the
mutated sequences inserted into the introns that are
still recognized as snoRNAs. As the mutations are
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
178

Figure 3: (a) Example of a mutation tree with 3 children and
a maximum of 5 nodes per level. The sequences shown in
the tree are in the positive set. (b) Example of a negative set
(sequences are not in the tree) and a positive set with 10%
mutated positions (10 nucleotide length sequence).
cumulative, the positive set comprises of sequences
with approximately the same percentage of mutations.
For example 10%, 20 %, 30%, 40% and 50% of po-
sitions mutated relative to the representative snoRNA
sequence. The negative set is formed by the mutated
sequences sMutated that could no longer be identi-
fied as snoRNAs. Each negative set is composed of
sequences with the same mutation percentage, simi-
lar to the positive set. For a better understanding of
the positive and negative sets, we illustrate an exam-
ple with 10% of mutated positions in Figure 3.
2.3 Feature Extraction
We extracted features for each sequence of positive
and negative sets obtained from the mutation tree.
(Fig 2-Step 4). The following features are extracted
from the C/D box sequences: mfe (Minimum Free En-
ergy of the secondary structure without constraints);
mfeC (MFE of the secondary structure with con-
straints); E
avg
(MFE average); E
stdv
(MFE standard
deviation); ls (length of the terminal stem); Dcd (dis-
tance between C and D boxes); C
score
(score of the
C box); D
score
(score of the D box); GC (GC con-
tent); zscore (z-score obtained by RNAz 2.0 (Gruber
et al., 2010)); bpStem (number of base pairs on the
terminal stem); lu5 (number of unpaired nucleotides
inside the stem before C box); lu3 (number of un-
paired nucleotides inside the stem after D box); stemU
npCbox (number of unpaired nucleotides between the
stem and the C box); stemU npDbox (number of un-
paired nucleotides between the D box and the stem).
The following features are extracted from the
H/ACA box sequences with snoReport 2.0: mfeC
(MFE of the secondary structure with constraints);
AC, GU, GC (AC, GU and GC content); zscore (zs-
core computed by RNAz); Hscore (score of the H
Table 1: C/d box and H/ACA box.
snoRNA Type tree N n(1) n(0)
substitution 3000 2574 2574
C/D insertion 3000 1795 1795
deletion 3000 2232 2232
substitution 2000 1835 1835
H/ACA insertion 2000 1009 1009
deletion 2000 1664 1664
box); ACAscore (score of the ACA box); LseqSize
(number of nucleotides before the H box); RseqSize
(number of nucleotides between H and ACA boxes);
LloopSC (lenght of the loop, where we find the pocket
region containing the target region, near to the H box);
RloopSC (length of the loop, where we find the pocket
region containing the target region, more close to the
ACA box); LloopY C (symmetry of the loop contain-
ing the pocket region near to the H box); RloopY
C (symmetry of the loop containing the pocket re-
gion near to the ACA box); LloopSym (symmetry of
all loops before H box); RloopSym (symmetry of all
loops before ACA box).
The datasets are files in Comma-separated values
(CSV) format composed of the same number of in-
stances extracted from the positive (1) and negative
(0) sets. Since different sequence may produce the
same feature values. Therefore, duplicated feature
vectors are removed from both the positive and nega-
tive set. For each mutation tree, we verify the pos-
itive set with the least number n of instances. We
constructed datasets comprising different numbers n
of sequences. For a given n, we randomly chose n in-
stances from the positive and negative sets according
to the percentage of mutation. Example, the dataset
with 10% mutation is composed of n instances of the
positive set with 10% mutation and n instances of the
negative set with approximately 10% mutation.
Normalization by linear interpolation (Gold-
schmidt and Passos, 2005) was applied to the features
extracted from the sequences of the positive and nega-
tive sets, i.e., feature values were transformed accord-
ing to x
0
= (x − x
min
)/(x
max
− x
min
). This preserves
proportional distances between normalized data and
the distances between the original data (Fig 2-Step 5).
For both C/D box and H/ACA box snoRNAs we gen-
erated three mutation trees, one for each of the types
of mutation (substitution, insertion, deletion). Table 1
list, for C/D box and H/ACA box, respectively, the
number N of sequences of the positive sets (Fig 2 -
Step 3), the number n(1) of feature vectors extracted
from the N sequences of the positive set, the number
n(0) of feature vectors extracted from the sequences
of the negative set (Fig 2 - Step 4).
Machine Learning Studies of Non-coding RNAs based on Artificially Constructed Training Data
179

2.4 Evaluation of Machine Learning
Methods
Our goal is to evaluate the use of ML methods in
the context of ncRNA homology search. To this
end, we apply three ML algorithms, SVM (Rus-
sell and Norvig, 2010), Artificial Neural Net-
work (ANN) (Haykin, 1999) and Random For-
est (RF) (Breiman, 2001). These methods were
chosen since they have been extensively used for
ncRNA classification tasks (Georgakilas et al., 2020;
Achawanantakun et al., 2015). The evaluation of
SVM is not entirely fair in comparison to the other
methods, since snoReport 2.0 also uses an SVM,
albeit trained on different data and embedded in ad-
ditional filters. It still provides valuable informa-
tion on the limitations of the ML approaches. We
use Jupyter-notebook to implement the supervised
learning algorithms, language packages in Python,
Keras (Gulli and Pal, 2017) and Scikit-learn (Pe-
dregosa et al., 2011). For SVM, we use the Ra-
dial basis function kernel (RBF), while for ANN we
use the sequential model of neural network, with
three layers of the Dense type, and, finally, for RF,
we use one hundred decision trees built using the
Bagging technique (by default). We tested all the
datasets on the ML algorithms, with 10-fold cross-
validation (Fig 2-Step 6). To evaluate ML algorithms,
we report the following evaluation metrics: Area Un-
der The Curve (AUC), Matthews Correlation Coeffi-
cient (MCC), Recall, Precision and Receiver Operat-
ing Characteristic Curve (ROC curve).
3 RESULTS AND DISCUSSION
We executed Blastn with default scoring using as
queries, the biological snoRNA sequences (tree roots)
and as databases each of the positive sets generated by
the mutation trees (with mutation rates of 10%, 20%,
30%, 40% and 50%, and N = 3, 000 for C/D box and
N = 2,000 for the H/ACA box). The sequences, the
original snoRNAs and the mutated ones, were tested
inside the introns, and also considering only the se-
quences themselves. To quantify the success of this
sequence-based homology search, we computed an
average hit rate, M := S/N, where S is the number
of aligned sequences and N is the total number of se-
quences (query file length).
With the snoRNAs inside the intron, Blastn al-
ways found matches, so in this case M = 100%. How-
ever, in most cases these did not match only the target
snoRNA but were spurious hits elsewhere in the in-
tron. Disregarding the decoy sequences, we obtained
essentially the same results with Blastn, independent
of the mutation model.
With the snoRNAs themselves, Blastn found
matches only for 10% and 20%, all of them with an
e-value ≤ 0.01. For substitutions, as example, we ob-
tained M = 18.9% and M = 0.5% for C/D box snoR-
NAs, and M = 73.6% and M = 2.9% for H/ACA box
snoRNAs at 10% and 20% mutation rate, respectively.
At even higher level of sequence divergence, Blastn
did not recover any snoRNA. We therefore have con-
structed a homology search problem that is very dif-
ficult for Blastn, the most widely used tool for this
task. These results certainly could be improved by
adapting Blastn for distance homologies or by using
Hidden Markov Models (HMMs) that use a pattern
instead of a single sequence as query. We did not pur-
sue this, since our main interest is to demonstrate that
ML algorithms are capable of handling such a diffi-
cult homology search problem.
3.1 C/D Box
We performed experiments for each mutation tree
(substitution, insertion and deletion). From these, we
chose substitution and insertion to discuss results in
more detail.
Substitution. Table 2 shows the results of the three
ML algorithms for the substitution experiment.
The ANN and SVM showed decreasing values for
all evaluation metrics. With 10% of ANN mutation
they obtained MCC = 98.84(%) and SVM MCC =
96.97(%), with 50% of mutation ANN and SVM they
obtained MCC = 63.36(%) and MCC = 44.44(%), re-
spectively. We can see in Fig 4 the ROC curves of
these two classifiers, the models achieved a good mea-
Table 2: Results ML algorithms for substitution with all fea-
tures C/D box: Area Under The Curve (AUC), Matthews
Correlation Coefficient (MCC), Recall (R) and Precision
(P).
ML Datasets AUC(%) MCC(%) R(%) P(%)
10(%) 99.42 98.84 99.18 99.65
20(%) 97.79 95.64 96.16 99.40
ANN 30(%) 93.93 88.65 88.35 99.43
40(%) 93.96 88.55 90.17 97.56
50(%) 80.32 63.36 72.27 86.16
10(%) 98.47 96.97 97.71 99.21
20(%) 96.14 92.35 94.49 97.71
SVM 30(%) 93.55 87.31 93.55 93.55
40(%) 88.53 77.45 89.59 87.72
50(%) 71.47 44.44 77.90 69.03
10(%) 77.21 56.61 57.32 95.23
20(%) 91.19 82.96 83.30 98.89
RF 30(%) 72.98 47.40 47.38 97.13
40(%) 93.03 86.52 90.80 95.04
50(%) 78.40 57.63 71.15 83.23
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
180

Figure 4: ROC curves of the datasets with 10%, 20%, 40%
and 50% of mutations, for substitution, with all features.
sure of separability for datasets with 10%, 20%, 30%
and 40% mutation. However, for datasets with 50%
mutation, they showed less capacity for class separa-
tion. RF obtained good results in all evaluation met-
rics for 20% and 40% mutation. The other datasets
10%, 30% and 50% did not achieve good results, pre-
diction with MCC = 56.61(%), MCC = 47.40(%) and
MCC = 57.63(%) respectively.
In order to investigate cases where biological char-
acteristics are not known, we also tested the ML algo-
rithms with a reduced number of features, in this case
- zscore, ls, Dcd, GC, lu5, and lu3. The three classi-
fiers did not achieve good results in all datasets, or the
models did not show a good predictive capacity.
Insertion. Table 3 shows the results of the three ML
algorithms for the insertion experiment.
The SVM classifier presented a good prediction in
all datasets, with 10% of mutations AUC = 97.71%,
and 50% of mutations, AUC = 94.01%, as shown by
Table 3: Results ML algorithms for insertion with all fea-
tures C/D box: Area Under The Curve (AUC), Matthews
Correlation Coefficient (MCC), Recall (R) and Precision
(P).
ML Datasets AUC(%) MCC(%) R(%) P(%)
10(%) 82.17 65.42 64.87 99.23
20(%) 81.70 68.21 63.77 99.48
ANN 30(%) 78.55 59.77 57.41 99.52
40(%) 91.73 84.58 84.97 98.26
50(%) 92.26 85.20 86.97 97.26
10(%) 97.71 95.53 95.94 99.48
20(%) 96.71 93.58 94.16 99.24
SVM 30(%) 93.76 88.17 88.75 98.64
40(%) 94.27 88.97 89.64 98.77
50(%) 94.01 88.48 89.37 98.53
10(%) 53.83 9.61 9.69 82.86
20(%) 49.3 -3.63 1.17 31.34
RF 30(%) 49.72 0.53 2.67 45.28
40(%) 50.11 1.16 0.56 62.50
50(%) 54.3 4.88 9.52 90.96
Figure 5: ROC curves of the datasets with 10%, 20%, 40%
and 50% of mutations, for insertion with all features.
the ROC curves in Fig 5. The RF classifier model
did not achieve class separation capability in all the
datasets, with 10% of mutations, AUC = 53.83%, and
50% of mutations AUC = 54.30%, as shown in the
ROC curves in Fig 5. The ANN classifier presents a
good prediction for the datasets, with 40% of muta-
tions, AUC = 91.73% and 50%, AUC = 92.26%, as
shown by the ROC curves in Fig 5. In this experi-
ment, the insertion mutation may have evidenced C/D
box characteristics.
We also tested a reduced number of features for in-
sertion, in this case zscore, ls, Dcd, GC, lu5, and lu3.
The ANN and SVM classifiers achieved lower perfor-
mance but still remained functional. As with substi-
tution, these two experiments with insertion showed
that the set of features is very important for predict-
ing the C/D box by ML classifiers. If relevant biolog-
ical characteristics are not known, the performance of
the classifiers deteriorates, in particular for the more
distant homologs.
3.2 H/ACA Box
We performed our experiments for each mutation
tree(substitution, insertion and deletion). From our
experiments, we choose substitution and insertion to
discuss results in more detail.
Substitution. Table 4 shows the results of the three
ML algorithms for the substitution experiment.
The SVM and RF classifiers showed decreasing
values for all evaluation metrics table 4. With 10%
of mutations, ANN obtained AUC = 74.20%, SVM
AUC = 67.76% and RF AUC = 62.03%. With 50%
mutation, the results were even lower, ANN, SVM
and RF obtained AUC = 59.08%, AUC = 61.36% and
AUC = 50.57% respectively. We can see the corre-
sponding ROC curves in Fig 6.
For H/ACA box, in order to investigate cases
Machine Learning Studies of Non-coding RNAs based on Artificially Constructed Training Data
181
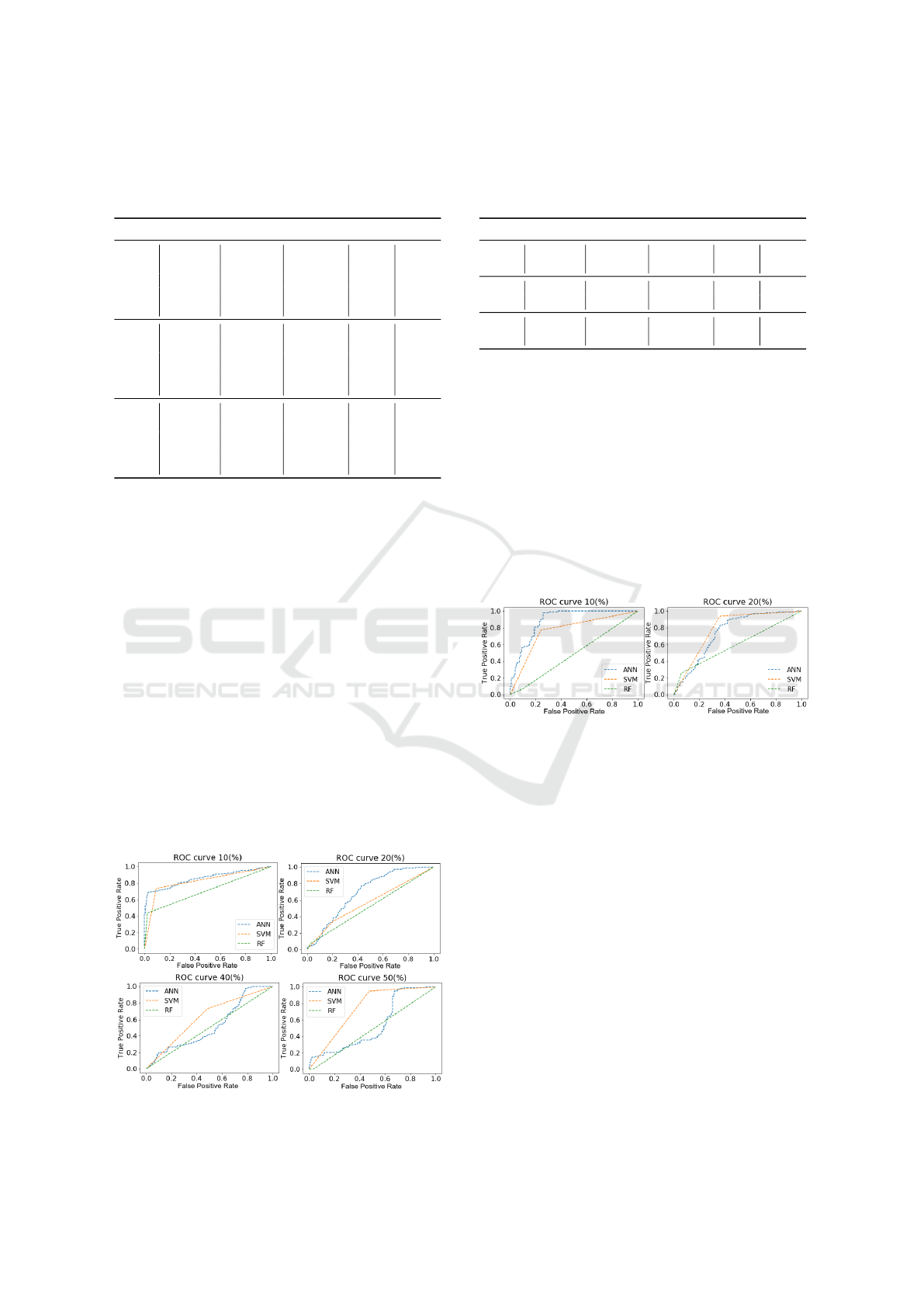
Table 4: Results ML algorithms for substitution with all
features H/ACA box: Area Under The Curve (AUC),
Matthews Correlation Coefficient (MCC), Recall (R) and
Precision (P).
ML Datasets AUC(%) MCC(%) R(%) P(%)
10(%) 74.20 50.03 71.08 75.83
20(%) 63.22 30.02 40.41 74.35
ANN 30(%) 64.66 32.98 51.53 69.92
40(%) 58.14 22.71 21.41 80.70
50(%) 59.08 23.15 29.41 72.29
10(%) 67.76 36.37 55.12 73.76
20(%) 66.20 33.29 54.9 70.99
SVM 30(%) 64.33 29.23 65.63 63.99
40(%) 61.34 24.17 70.53 59.60
50(%) 61.36 22.78 54.85 63.06
10(%) 62.03 28.86 27.51 88.75
20(%) 52.75 2.38 17.43 59.37
RF 30(%) 60.64 21.59 29.47 78.29
40(%) 52.42 7.19 6.81 77.64
50(%) 50.57 2.92 4.52 57.24
where biological characteristics are not known, we
also tested the ML algorithms with a reduced number
of features, in this case zscore, AC, GU, GC, LloopSC,
RloopSC, LloopYC, and RloopYC. The three classi-
fiers performed poorly on all datasets, with an AUC
below 65%.
Insertion. Table 5 shows the results of the three ML
algorithms for the insertion experiment. In this ex-
periment, the mutation tree only generated sequences
recognized as H/ACA box by snoReport 2.0 up to
20% mutation. We could observe that the length of
the snoRNA sequence is an important feature for its
identification.
The three classifiers presented a performance with
10% mutation higher than with 20% mutation. How-
ever, all metrics are close to or below 70%, see fig 7.
Only ANN and SVM showed AUC and Recall greater
than 70% with 10% mutation. The results of the
three classifiers with 20 % mutation corresponding
Figure 6: ROC curves of the datasets with 10%, 20%, 40%
and 50% of mutation, for substitution, with all features.
Table 5: Results ML algorithms for insertion with all
features H/ACA box: Area Under The Curve (AUC),
Matthews Correlation Coefficient (MCC), Recall (R) and
Precision (P).
ML Datasets AUC(%) MCC(%) R(%) P(%)
10(%) 71.92 47.90 59.31 79.3
ANN 20(%) 59.30 19.80 45.05 63.02
10(%) 74.76 51.45 80.4 72.31
SVM 20(%) 62.1 25.13 69.01 60.66
10(%) 59.79 22.83 28.91 75.65
RF 20(%) 51.21 1.81 28.32 52.29
to less than ideal performance. Again, we tested the
ML techniques with a reduced number of features:
zscore, AC, GU, GC, LloopSC, RloopSC, LloopYC,
and RloopYC. All three classifiers performed better
with 10% of mutations than with 20%, with the per-
formances decreasing further with the increasing of
number of mutations. For H/ACA box, the perfor-
mance of the AM classifiers were equivalent, using a
large number of known biological features as well as
a small number of them. For the insertion mutation,
the higher the percentage of mutation, the worse the
performance of the three classifiers.
Figure 7: ROC curve of the datasets 10% and 20%, for in-
sertion with all features.
4 CONCLUSION
In this article, we studied the performance of ML
methods to predict snoRNAs with the help of large
datasets built from artificially constructed mutation
trees. Even with limitations, we found that the ML
methods performed better than the most common con-
ventional homology search, Blast, which considers
only sequence similarity. As expected, the Blast re-
sults showed many false negative results for snoRNAs
with low sequence similarity to the query. For C/D
box, we observed that the ML methods consistently
performed better when provided with all known bi-
ologically relevant features. This was in particular
the case for the most diverged sequences. For the
substitution experiment, the SVM and ANN classi-
fiers achieved excellent performance for datasets with
10%, 20%, 30%, and 40% of mutations. A large drop
in performance was observed for 50% of mutations.
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
182

For H/ACA box, the performance of the ML classi-
fiers, both using the full set of known biological char-
acteristics and a reduced number of features, showed
equivalent prediction performance. In the experiment
with the insertion mutation, performance decreased
with increasing mutation levels for all the three ML
classifiers.
In summary, our results show that ML methods
can be competitive with traditional homology search
methods, provided sufficiently large sets of indepen-
dent instances for test and training sets. This require-
ment, however, is prohibitive for most practical appli-
cations. We therefore suggest that the careful produc-
tion of artificial data is a promising approach that can
be pursued in practice, at least for families of ncRNAs
for which an adequate diverse set of representatives is
available. Our data also indicate that the knowledge
of a sufficiently large set of biologically relevant fea-
tures is important for the performance of ML-based
homology search.
Clearly, the present study is only a first step.
It remains open whether the ML methods can also
compete with more sophisticated methods of homol-
ogy search such as Hidden Markov Models (HMMs)
(Eddy, 1996) or covariance models (CMs) (Nawrocki
and Eddy, 2013), which similar to ML models also
convey information of local and non-local correla-
tions, respectively.
REFERENCES
Achawanantakun, R., Chen, J., Sun, Y., and Zhang, Y.
(2015). LncRNA-ID: Long non-coding RNA IDentifi-
cation using balanced random forests. Bioinformatics,
31(24):3897–3905.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and
Lipman, D. J. (1990). Basic local alignment search
tool. J. Mol. Biol., 215(3):403–410.
Barber, D. (2012). Bayesian Reasoning and Machine
Learning. Cambridge University Press, Cambridge,
UK.
Bartschat, S., Kehr, S., Tafer, H., Stadler, P. F., and Hertel,
J. (2014). snoStrip: a snoRNA annotation pipeline.
Bioinformatics, 30(1):115–116.
Bratkovi
ˇ
c, T., Bo
ˇ
zi
ˇ
c, J., and Rogelj, B. (2020). Functional
diversity of small nucleolar RNAs. Nucleic Acids Re-
search, 48(4):1627–1651.
Breiman, L. (2001). Random forests. Machine Learning,
45:5–32.
de Araujo Oliveira, J. V., Costa, F., Backofen, R., Stadler,
P. F., Machado Telles Walter, M. E., and Hertel, J.
(2016). SnoReport 2.0: new features and a refined
Support Vector Machine to improve snoRNA identifi-
cation. BMC Bioinformatics, 17 Suppl. 18:464.
Eddy, S. R. (1996). Hidden Markov models. Current Op.
Struct. Biol., 6:361–365.
Falaleeva, M. and Stamm, S. (2013). Processing of snoR-
NAs as a new source of regulatory non-coding RNAs:
snoRNA fragments form a new class of functional
RNAs. BioEssays, 35(1):46–54.
Georgakilas, G. K., Grioni, A., Liakos, K. G., Chalupova,
E., Plessas, F. C., and Alexiou, P. (2020). Multi-
branch Convolutional Neural Network for Identifica-
tion of Small Non-coding RNA genomic loci. Scien-
tific Reports, 10(1):9486.
Goldschmidt, R. and Passos, E. (2005). Data mining: a
Practical guide. Gulf Professional Publishing.
Gruber, A. R., Findeiß, S., Washietl, S., Hofacker, I. L., and
Stadler, P. F. (2010). RNAz 2.0: improved noncoding
RNA detection. Pac. Symp. Biocomput., 15:69–79.
Gulli, A. and Pal, S. (2017). Deep Learning with Keras.
Packt Publishing Ltd, Birmingham, UK.
Haykin, S. (1999). Neural Networks: A Comprehensive
Foundation. Prentice-Hall, Englewood Cliffs.
Lorenz, R., Bernhart, S. H., H
¨
oner zu Siederdissen, C.,
Tafer, H., Flamm, C., Stadler, P. F., and Hofacker, I. L.
(2011). ViennaRNA Package 2.0. Alg. Mol. Biol.,
6:26.
Nawrocki, E. P. and Eddy, S. R. (2013). Infernal 1.1: 100-
fold faster RNA homology searches. Bioinformatics,
29(22):2933–2935.
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V.,
Thirion, B., Grisel, O., Blondel, M., Prettenhofer, P.,
Weiss, R., Dubourg, V., Vanderplas, J. T., Passos, A.,
Cournapeau, D., Brucher, M., Perrot, M., and Duch-
esnay,
´
E. (2011). Scikit-learn: Machine learning in
python. J. Machine Learning Res., 12:2825–2830.
Rastogi, A. and Gupta, D. (2014). GFF-Ex: a genome fea-
ture extraction package. BMC research notes, 7:315.
Russell, S. and Norvig, P. (2010). Artificial Intelligence: A
Modern Approach. Prentice-Hall, Englewood Cliffs,
3nd edition.
Satoh, N. (2003). The ascidian tadpole larva: compara-
tive molecular development and genomics. Nature Re-
views Genetics, 4(4):285–295.
Waldl, M., Thiel, B., Ochsenreiter, R., Holzenleiter, A.,
de Araujo Oliveira, J. V., Walter, M. E. M. T., Wolfin-
ger, M. T., and Stadler, P. F. (2018). TERribly difficult:
Searching for telomerase RNAs in Saccharomycetes.
Genes, 9:372.
Zhang, Y., Huang, H., Zhang, D., Qiu, J., Yang, J., Wang,
K., Zhu, L., Fan, J., and Yang, J. (2017). A Review on
Recent Computational Methods for Predicting Non-
coding RNAs. BioMed Res. Intl., 2017:1–14.
Zhang, Y. and Rajapakse, J. C. (2009). Machine Learning
in Bioinformatics. John Wiley & Sons, Hoboken, NJ.
Machine Learning Studies of Non-coding RNAs based on Artificially Constructed Training Data
183
