
A Deep Learning Method to Impute Missing Values and Compress
Genome-wide Polymorphism Data in Rice
Tanzila Islam
1a
, Chyon Hae Kim
1b
, Hiroyoshi Iwata
2c
, Hiroyuki Shimono
3d
, Akio Kimura
1
,
Hein Zaw
4
, Chitra Raghavan
4
, Hei Leung
4
and Rakesh Kumar Singh
5e
1
Department of Systems Innovation Engineering, Graduate School of Science and Engineering, Iwate University,
Morioka, Iwate, Japan
2
Department of Agricultural and Environmental Biology, Graduate School of Agricultural and Life Sciences,
The University of Tokyo, Bunkyo, Tokyo, Japan
3
Crop Science Laboratory, Faculty of Agriculture, Iwate University, Morioka, Japan
4
International Rice Research Institute (IRRI), Laguna, Philippines
5
International Center for Biosaline Agriculture (ICBA), Dubai, U.A.E.
kimura@cis.iwate-u.ac.jp, heinzawagri@gmail.com, chitrarag2006@gmail.com, hleung1155@gmail.com,
r.singh@biosaline.org.ae
Keywords: Genome-wide DNA Polymorphisms, Stacked Autoencoder, Deep Neural Network, Separate Stacking Model,
Genome Compression, Missing Value Imputation.
Abstract: Missing value imputation and compressing genome-wide DNA polymorphism data are considered as a
challenging task in genomic data analysis. Missing data consists in the lack of information in a dataset that
directly influences data analysis performance. The aim is to develop a deep learning model named
Autoencoder Genome Imputation and Compression (AGIC) which can impute missing values and compress
genome-wide polymorphism data using a separated neural network model to reduce the computational time.
This research will challenge the construction of a model by using Autoencoder for genomic analysis, in other
words, a fusion research between agriculture and information sciences. Moreover, there is no knowledge of
missing value imputation and genome-wide polymorphism data compression using Separated Stacking
Autoencoder Model. The main contributions are: (1) missing value imputation of genome-wide
polymorphism data, (2) genome-wide polymorphism data compression of Rice DNA. To demonstrate the
usage of AGIC model, real genome-wide polymorphism data from a rice MAGIC population has been used.
1 INTRODUCTION
Missing value imputation and genome-wide
polymorphism data compression have an important
role in genomic data analysis. Reducing the
dimension of genome-wide polymorphisms data
minimizes the calculation time. The general purpose
of this study is to develop a Deep Learning model
called Autoencoder Genome Imputation and
Compression (AGIC) which has a function for
a
https://orcid.org/0000-0002-3203-4083
b
https://orcid.org/0000-0002-7360-4986
c
https://orcid.org/0000-0002-6747-7036
d
https://orcid.org/0000-0002-7328-0483
e
https://orcid.org/0000-0001-7463-3044
imputing missing values as well as compressing
genome-wide polymorphism data at a time.
Deep learning has emerged from machine
learning methods inspired by artificial neural
networks with hierarchical feature learning. It
enhances analysis accuracy, and data analysis can be
performed with higher accuracy than conventional
methods. Deep Learning architectures like deep
neural networks, belief networks, and recurrent
neural networks, and convolutional neural networks
have found applications in the field of computer
Islam, T., Kim, C., Iwata, H., Shimono, H., Kimura, A., Zaw, H., Raghavan, C., Leung, H. and Singh, R.
A Deep Learning Method to Impute Missing Values and Compress Genome-wide Polymorphism Data in Rice.
DOI: 10.5220/0010233901010109
In Proceedings of the 14th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2021) - Volume 3: BIOINFORMATICS, pages 101-109
ISBN: 978-989-758-490-9
Copyright
c
2021 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
101

vision, audio or speech recognition, machine
translation, social network filtering, bioinformatics
(Li et al., 2019) and other diverse fields. Recently it
has been introduced in the field of agriculture
(Kamilaris & Prenafeta-Boldú, 2018). Deep Learning
provides many advanced methods, one of which is
autoencoder. An auto-encoder (Gulli & Pal, 2017) is
a type of neural network that can learn a compressed
and distributed representation (i.e. encoding) of the
input data, thus it can be used for dimensionality
reduction.
The main contributions of this paper are:
1. To develop a new method for imputing
missing values with an autoencoder based
on the Deep Neural Network. This method
demonstrates that by using an autoencoder,
it is possible to achieve accurate imputation
of missing values in genome-wide
polymorphism data.
2. To develop a genome compression method
by using Separated Stacking Autoencoder.
This compression method is scalable for a
large number of genome-wide
polymorphisms, and beneficial for saving
storage as well as computational time.
The remainder of this paper is structured as
follows: In Section 2, a review of some literatures of
missing value imputation and genome-wide
polymorphism data compression in the domain of
bioinformatics is discussed. Section 3 introduces the
pipeline of missing value imputation and genome
compression method AGIC. The pipeline of AGIC
method includes the pre-processing of genome-wide
polymorphism data, the autoencoder training for
missing value imputation as well as genome-wide
polymorphism data compression. Section 4 evaluates
the result by comparing the performance of existing
methods and AGIC, and finally conclusion is drawn
in Section 5.
2 LITERATURE REVIEW
It is a challenging task to compress genome-wide
polymorphism data (Grumbach & Tahi, 1994) and
impute missing values in the data effectively
(Troyanskaya et al., 2001). Since the last decade,
researchers have been exploring various approaches
to compress genome-wide polymorphism data (Wang
et al., 2018) and impute missing values (Gad,
Hosahalli, Manjunatha, & Ghoneim, 2020; Rana,
John, & Midi, 2012).
There have been many studies addressing the
traditional genotype imputation methods which are
typically based on haplotype-clustering algorithms
(Scheet & Stephens, 2006) and hidden Markov
models (Marchini, Howie, Myers, McVean, &
Donnelly, 2007). BEAGLE is another imputation tool
based on a graphical model of a set of haplotypes
(Browning & Browning, 2009; Browning, Zhou, &
Browning, 2018). It works iteratively by fitting the
model to the current set of estimated haplotypes. Then
resampling of new estimated haplotypes for each
individual is conducted based on a fitted model. The
probabilities of missing genotypes are calculated
from the fitted model at the final iteration. Recently,
deep learning based methods, especially
autoencoders have shown great potential to impute
missing values (Beaulieu-Jones & Moore, 2017;
Duan, Lv, Liu, & Wang, 2016); for example Abdella
and Marwala proposed a method to approximate
missing data by using an autoencoder and genetic
algorithm (Abdella & Marwala, 2005).
In 2018, Lina proposed a Denoising Autoencoder
with Partial Loss (DAPL) method to predict missing
values in pan-cancer genomic analysis (Qiu, Zheng,
& Gavaert, 2018). They showed that the DAPL
method achieves better performance with less
computational burden over traditional imputation
methods.
In 2019, Chen and Shi proposed a deep model
called a Sparse Convolutional Denoising
Autoencoder (SCDA) to impute missing values of
human and yeast genotype data respectively (Chen &
Shi, 2019). This SCDA model achieves significant
imputation accuracy compared with popular
reference-free imputation methods.
In 2020, Lina proposed a deep learning
imputation framework for transcriptome and
methylome data using a Variational AutoEncoders
(VAE) and showed that it can be a preferable
alternative to traditional methods for data imputation,
especially in the setting of large-scale data and certain
missing-not-at-random scenarios (Qiu, Zheng, &
Gevaert, 2020).
On the other hand, there have been many research
addressing genome compression based on neural
networks. In 2006, Hinton and Salakhutdinov
proposed a method based on neural networks by using
a Restricted Boltzmann Machine (RBM) to reduce the
binary stochastic information and dimensions of the
data (Hinton & Salakhutdinov, 2006). In 2016, Sento
introduced a method for image compression using an
autoencoder (Sento, 2016). The accuracy of this
method is high, and results showed that Deep Neural
Network (DNN) could be efficient for image
compression purposes.
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
102
In 2019, Wang and Zang proposed DeepDNA to
compress human mitochondrial genome data using
machine learning techniques. The method has a good
compression result in the population genome with
large redundancy, and in the single genome with
small redundancy (Wang, Zang, & Wang, 2019).
As a recent paper, Absardi and Javidan introduced
a fast reference free genome compression using
Autoencoder to reduce the compression time and to
keep the compression ratio on an acceptable level
(Absardi & Javidan, 2019).
Above mentioned works have discussed either
missing value imputation, or genome-wide
polymorphism data compression individually. There
is no study has been done to impute missing values
and compress genome-wide polymorphisms data at a
time using a separated network. Hence in this study,
a deep learning method Autoencoder Genome
Imputation and Compression (AGIC) has been
introduced which can impute missing values and
compress genome-wide polymorphism data at a time
using a separated stacked autoencoder.
3 METHODS
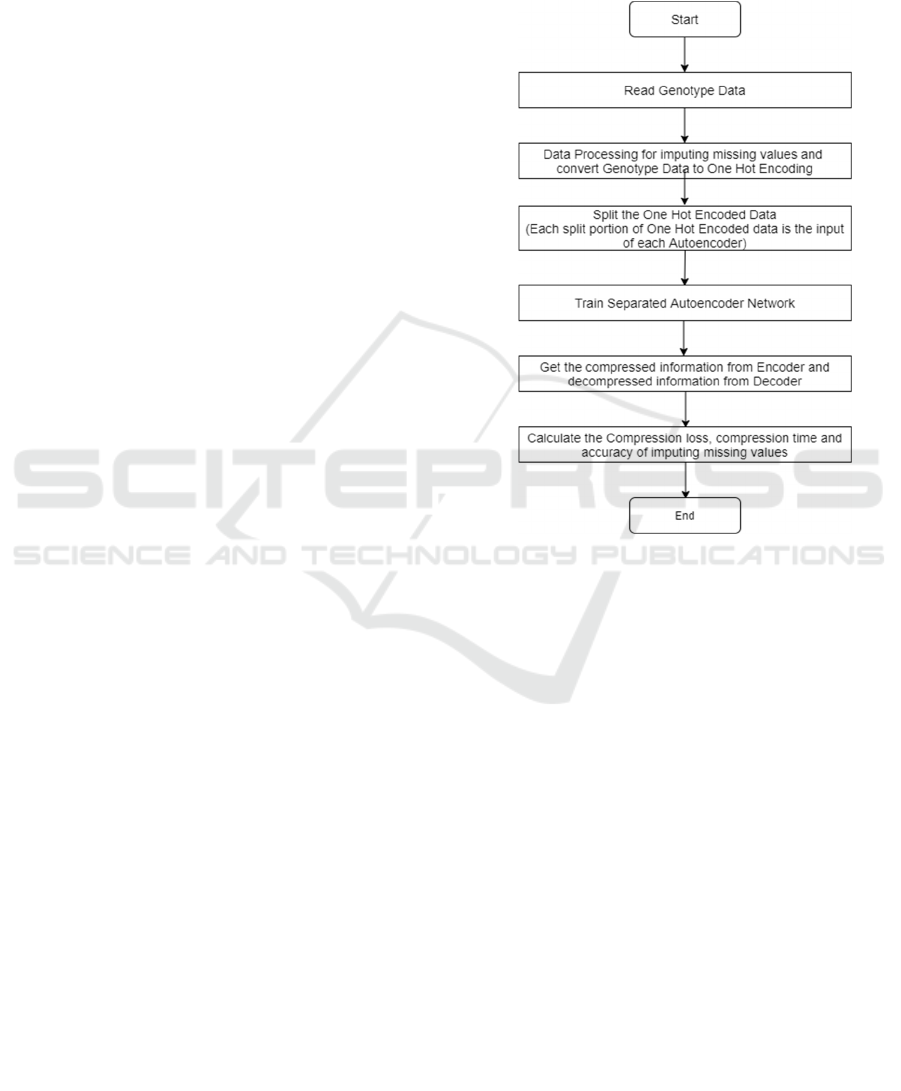
A deep learning method AGIC comprises a few steps
which are shown in Figure 1. In the first step, the
genome-wide polymorphism data (genotype data) has
been read. Therefore, the genotype data has been pre-
processed by imputing missing values and then
converted into one hot encode. After processing the
categorical values through one hot encoding, an input
data splitting technique has been performed to reduce
the calculation time. Then, an autoencoder model has
been trained by taking each split portion of data as an
input. By training the autoencoder, the compression
and decompression have been done with encoder and
decoder, respectively. Finally, the model has been
evaluated by calculating the compression loss,
compression time and accuracy of imputing missing
values.
All experiments in this study have been conducted
on a PC with an Intel(R) Core(TM) i9-9900K, 3.60
GHz CPU, 32 GB RAM and 64 bit Windows 10
operating system.
3.1 Dataset
The genotype data used in this study was obtained
from a multiparent advanced generation intercross
(MAGIC) population derived from eight indica rice
varieties (Fedearroz 50, Shan-Huang Zhang-2,
IRRI123, IR77186-122-2-2-3, IR77298-14-1-2-10,
IR4630-22-2-5-1-3, IR45427-2B-2-2B-1-1, Sambha
Mahsuri + Sub1). The dataset comprises genotypes of
genome-wide polymorphisms of 1,316 lines for
27,041 SNPs in an excel file. This dataset contains 12
chromosomes and 4 bases of Rice DNA A (Adenine),
C (Cytosine), G (Guanine) and T (Thymine).
Figure 1: The flowchart of Autoencoder Genome
Imputation and Compression (AGIC).
3.2 Data Pre-processing
Data preprocessing plays an important role in
machine learning and deep learning algorithms, and
proper preprocessing of the data is compulsory for
achieving better performance. Machine learning and
deep learning models, like those in Keras, require all
input and output variables to be numeric. This means
that if the data contains categorical data, it is
mandatory to encode it to numbers before fitting and
evaluating a model. Therefore, a pre-processing
method is required to represent the categorical values
into numerical values. Pre-processing has been done
by applying the following steps:
3.2.1 One Hot Encoding
One hot encoding has been used to represent the
categorical values into numerical values. In this
strategy, each category value is converted into a new
column and assigned a 1 or 0 (notation for true/false)
A Deep Learning Method to Impute Missing Values and Compress Genome-wide Polymorphism Data in Rice
103

value to the column. The genotype data contains
categorical values such as A (Adenine), C (Cytosine),
G (Guanine) and T (Thymine).
Figure 2: Data Pre-processing (One Hot Encoding).
The datasets may have missing values, this can
cause problems while training a model. In Python,
One Hot Encoding can be done using sci-kit learn
library approach or using dummies values approach.
In this study, a dummy values approach has been used
for one hot encoding. All genomes are encoded into
one hot encoding by a 4-bit coding scheme: “A”, “C”,
“G” and “T” are encoded by “1000”, “0100”, “0010”
and “0001”, respectively. Figure 2 shows an example
of one hot encoding which has applied some
sequences of genotypes.
3.2.2 Splitting Input Data
After processing the raw data (1316 x 27041) through
One Hot Encoding, the encoded data is transformed
into a Python NumPy array. Now the shape of data
has become (1316 x 108164). As the shape of input
data is large, the input data splitting technique has
been applied to reduce the computational time at a
time. By considering 3863 to be the number of splits,
numpy hsplit has been used to split the one hot encode
array horizontally (axis =1 i.e. 108164). Each split
contains (1316 x 28) of data i.e. an input layer with
28 neurons in each network. The next sections are
devoted to explaining the autoencoder training by
considering the splitted input data.
3.3 Autoencoder Training
In this study a deep autoencoder has been used, which
is composed of two symmetrical deep-belief
networks that typically have three or four shallow
layers representing the encoding half of the network,
and a second set of three or four layers represents the
decoding half. Figure 3 shows the basic structure of
Deep Autoencoder.
Figure 3: Basic structure of Deep Autoencoder.
Given a set of training samples 𝑥
,𝑥
,𝑥
,...,
where 𝑥
∈ 𝑅
, an autoencoder first encodes an input
𝑥
to a hidden representation ℎ𝑥
based on (1), and
then it decodes representation ℎ𝑥
back into a
reconstruction 𝑥′𝑥
computed as in (2), as shown in:
ℎ𝑥
𝑓
𝑊𝑥 𝑏
(1)
𝑥′𝑥 𝑔𝑊′ℎ𝑥 𝑐
(2)
where 𝑓 is an activation function, 𝑊 is a weight
matrix, 𝑏 is an encoding bias vector, 𝑔 is a decoding
activation function, 𝑊′ is a decoding matrix, and 𝑐 is
a decoding bias vector.
The activation function of each layer except the
decoder layer is “ReLU” which stated in (3):
𝑓
𝑥 𝑚𝑎𝑥𝑥, 0
(3)
The activation function of the decoder layer is
“sigmoid” which is shown in (4):
𝑔𝑥 1/1 𝑒
(4)
The model has been implemented using Keras
Functional API, built on top of Tensorflow. The deep
structure of a network includes 9 layers. There are 2
layers in both the encoder and decoder without
considering the input and output. The number of
nodes per layer decreases with each subsequent layer
of the encoder and increases back in the decoder.
Also, the decoder is symmetric to the encoder in
terms of layer structure.
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
104

Figure 4: Signal flow graph of an autoencoder network built
on a Keras framework with the input and output dimension.
In Figure 4, the input layer of a network has 28
nodes, the first hidden layer has 14 nodes, the second
hidden layer has 7 nodes, and the code size is 3.
Figure 5: Training and Validation Loss of Deep
Autoencoder.
The model has been trained using an Adam
optimizer with the objective of minimizing the mean
squared error (MSE). 20 percent data has been used
to validate the experiment. Figure 5 shows the
training and validation loss of Deep Autoencoder.
4 RESULTS
4.1 Missing Value Imputation
The presence of missing values is a frequent problem
in the analysis of genome-wide polymorphism data.
In the original dataset, there was no missing value.
We simulated a range of missing proportions at 5%,
10%, 15% and 20% of the original data. The imputed
genotypes and true genotypes of the simulated
missing entries have been compared to find the
accuracy of missing value imputation. Four
approaches have been compared to validate the
imputation accuracy.
a.
Imputing Missing Values by AGIC (Replacing 0):
The missing value ‘N’ has been replaced by 0, while
converting the genome-wide polymorphisms to one
hot encodes. After training the autoencoder model,
the decoded genotypes were compared with true
genotypes of the simulated missing entries based on
their one hot encodes to calculate the accuracy of the
imputation.
Figure 6: The accuracy of imputing missing values by
AGIC (Replacing 0).
Figure 6 illustrates the accuracy of imputing
missing values by AGIC (Replacing 0). The accuracy
of imputation at the 5%, 10%, 15% and 20% missing
proportions are 94.15%, 88.37%, 81.00% and
72.04%, respectively.
b.
Imputing Missing Values by Simple Imputer (SI):
A simple statistical approach has been used to impute
missing values. In this approach, the missing value
has been replaced by the most frequent value at the
polymorphism and then converted the replaced value
into a one hot encode.
Figure 7: The accuracy of imputing missing values by
Simple Imputer (SI).
A Deep Learning Method to Impute Missing Values and Compress Genome-wide Polymorphism Data in Rice
105
The accuracy has been calculated before training
the network. Figure 7 shows the accuracy of imputing
missing values by Simple Imputer (SI). The accuracy
of imputation at the 5%, 10%, 15% and 20% missing
proportions are 92.48%, 86.17%, 80.85% and
76.44%, respectively.
c.
Imputing Missing Values by SI_AGIC:
The
autoencoder model has been trained by taking simple
imputed values as an input. After training the
autoencoder model, the decoded genotypes were
compared with true genotypes of the simulated
missing entries based on their one hot encode to
calculate the accuracy of the imputation.
Figure 8: The accuracy of imputing missing values by
SI_AGIC.
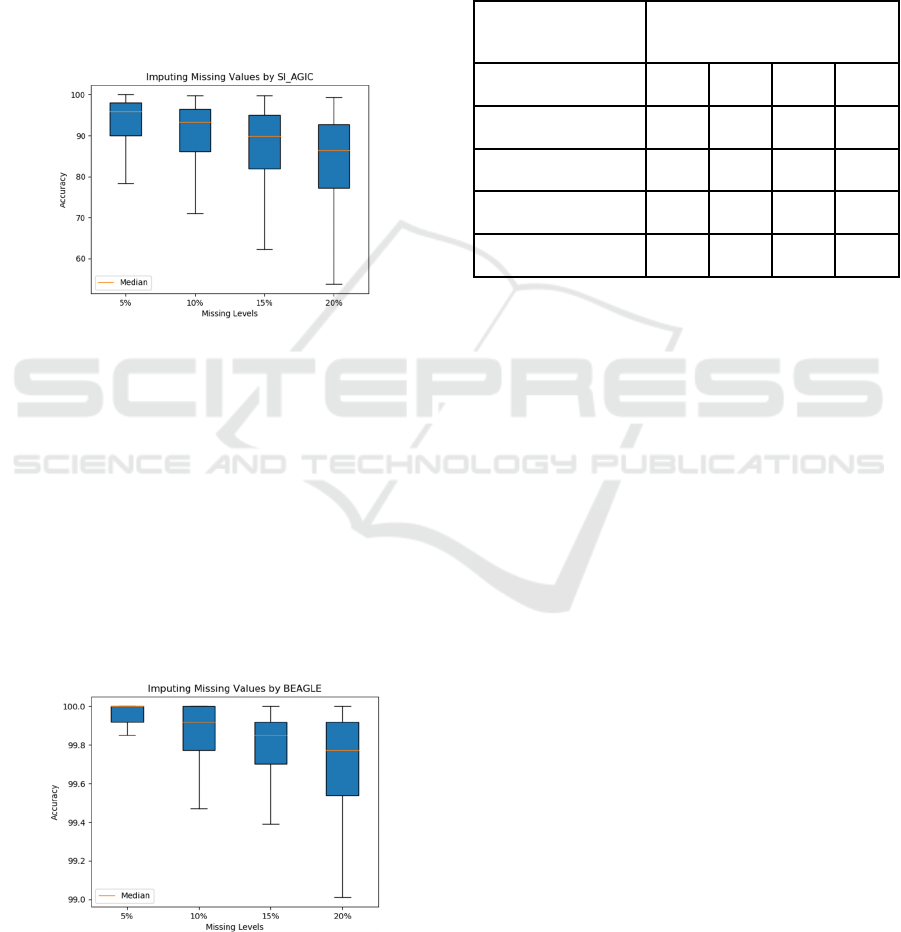
Figure 8 shows the accuracy of imputing missing
values by SI_AGIC. The accuracy of imputation at
the 5%, 10%, 15% and 20% missing proportions are
95.97%, 93.25%, 89.89% and 86.39%, respectively.
In this approach, the accuracy was better than the
other two approaches.
d.
Imputing Missing Values by BEAGLE:
A
common imputation method using BEAGLE, which
is a familiar program in genomic data analysis, was
applied. In this method, the missing genome data
were imputed by using BEAGLE 5.1 (Browning et
al., 2018).
Figure 9: The accuracy of imputing missing values by
BEAGLE.
Figure 9 shows the accuracy of imputing missing
values by BEAGLE. The accuracy of imputation at
the 5%, 10%, 15% and 20% missing proportions are
99.91%, 99.80%, 99.69% and 98.40%, respectively.
Table 1 shows the summary of missing
proportions and the accuracies of imputation
methods.
Table 1: Summary of Missing Proportions and the
Accuracies of Imputation Methods.
Methods Accuracy
5% 10% 15% 20%
AGIC (Replacing 0) 94.15 88.37 81.00 72.04
SI 92.48 86.17 80.85 76.44
SI
_
AGIC 95.97 93.25 89.89 86.39
BEAGLE 99.91 99.80 99.69 98.40
Although the accuracy of BEAGLE method was
higher than AGIC method, BEAGLE does not
provide any function for compression of genome-
wide polymorphism data. AGIC method enables
compress genome-wide polymorphism data and
impute missing values in the data at the same time.
4.2 Genome Compression
The requirement of data compression is the
dimensionality of the input and output needs to be the
same. The number of nodes in the middle layer is a
hyperparameter that gives the compressed
information. The signal flow graph of Gene Data
Compression with the input and output dimensions is
shown in Figure 10. A separate stacked autoencoder
has been used as a learning algorithm. The primary
reason to use a separate stacked autoencoder is to
reduce the computational time of the network.
4.2.1 Performance Evaluation
Since each autoencoder consists of an input layer, a
hidden layer, and an output layer, when implementing
the stacking process of the stacked autoencoder, the
input samples are sent to the input layer of the first
layer autoencoder first, and then these data in the
input layer are mapped to the hidden layer. Next, the
hidden layer data are mapped to the output layer.
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
106

Figure 10: Signal Flow Graph of Gene Data Compression.
After that, the value of the output layer and the
value of the input layer are used to calculate the
reconstruction error. The reconstruction error was
calculated as the mean squared error (MSE) function
shown in formula (5).
𝑀𝑆𝐸𝑥, 𝑥′ ||𝑥 𝑥′||
(5)
The MSE loss for compressing rice genome-wide
polymorphism data is 0.0078. The test time for
compressing genome data is 2244.89 sec.
4.2.2 Comparison with Other Compression
Methods
AGIC method was compared to one of a reference
free compression method which also used an
autoencoder to compress genome expressions of
Koref Dataset. (Absardi & Javidan, 2019).
The result of compressing Koref dataset by
considering AGIC method and Fast RefeTrence Free
method is presented below. In the Fast Reference Free
method, the input layer 𝑥 of a network has 15
neurons and the encoder layer ℎ has 3 neurons. The
Fast Reference Free model has been trained by
considering binary cross-entropy loss function. On
the other hand, in AGIC method, the input layer 𝑥
of a network has 28 neurons and the encoder layer ℎ
has 3 neurons. The loss has been calculated by using
mean squared error (MSE) in AGIC method. The
compression ratio was calculated as in formula (6).
𝐶𝑜𝑚𝑝𝑟𝑒𝑠𝑠𝑖𝑜𝑛 𝑅𝑎𝑡𝑖𝑜 𝑥/ℎ
(6)
Each training has been performed within 50
epochs considering an Adam optimizer. The
compression ratio, testing time and loss of Fast
Reference Free Method has been compared with
AGIC method in Table 2.
Table 2: Comparison of Fast Reference Free Method and
AGIC method by considering Koref Dataset.
Ratio Time Loss
Fast Reference Free
Metho
d
5 42.62 sec 0.2101
AGIC 9 8403.32 sec 0.2107
Though the compression time of AGIC is higher
than the Fast Reference Free method, the Fast
Reference Free method is not scalable to a large
number of genes. Because the calculation cost of Fast
Reference Free network will increase squared order
against the size of polymorphisms (sequences). On
the other hand, AGIC method is scalable for a large
number of genes, as a separated stacking network has
been considered. So, the calculation cost of AGIC
method will increase with linear order.
A Deep Learning Method to Impute Missing Values and Compress Genome-wide Polymorphism Data in Rice
107

5 CONCLUSIONS
In summary, a novel deep learning model AGIC as a
new paradigm was introduced to impute missing
values and compress genome expressions. The results
showed that AGIC model can achieve up to 96%
accuracy to impute missing values.
Moreover, this learning method is scalable for the
data of the large number of genome-wide
polymorphisms. A separate stacking model has been
implemented to minimize the calculation cost of the
network. The calculation cost of the network in AGIC
method increases with linear order, whereas
calculation costs of other popular methods increases
rapidly if the number of genome-wide
polymorphisms increases. AGIC model provides a
strong alternative to traditional methods for imputing
missing values and compressing genome expressions
at a time.
ACKNOWLEDGEMENTS
This research has been partially supported by the
JSPS KAKENHI (Grants-in-Aid for Scientific
Research) JP19H00938.
REFERENCES
Abdella, M., & Marwala, T. (2005). The use of genetic
algorithms and neural networks to approximate missing
data in database. IEEE 3rd International Conference on
Computational Cybernetics, 2005. ICCC 2005. (pp.
207–212). Presented at the IEEE 3rd International
Conference on Computational Cybernetics, 2005.
ICCC 2005., IEEE.
Absardi, Z. N., & Javidan, R. (2019). A Fast Reference-
Free Genome Compression Using Deep Neural
Networks. 2019 Big Data, Knowledge and Control
Systems Engineering (BdKCSE) (pp. 1–7). Presented at
the 2019 Big Data, Knowledge and Control Systems
Engineering (BdKCSE), IEEE.
Beaulieu-Jones, B. K., & Moore, J. H. (2017). Missing data
imputation in the electronic health record using deeply
learned autoencoders. Pacific Symposium on
Biocomputing, 22, 207–218.
Browning, B. L., & Browning, S. R. (2009). A unified
approach to genotype imputation and haplotype-phase
inference for large data sets of trios and unrelated
individuals. American Journal of Human Genetics,
84(2), 210–223.
Browning, B. L., Zhou, Y., & Browning, S. R. (2018). A
One-Penny Imputed Genome from Next-Generation
Reference Panels. American Journal of Human
Genetics, 103(3), 338–348.
Chen, J., & Shi, X. (2019). Sparse convolutional denoising
autoencoders for genotype imputation. Genes, 10(9).
Duan, Y., Lv, Y., Liu, Y.-L., & Wang, F.-Y. (2016). An
efficient realization of deep learning for traffic data
imputation. Transportation Research Part C: Emerging
Technologies, 72, 168–181.
Gad, I., Hosahalli, D., Manjunatha, B. R., & Ghoneim, O.
A. (2020). A robust deep learning model for missing
value imputation in big NCDC dataset. Iran Journal of
Computer Science.
Grumbach, S., & Tahi, F. (1994). A new challenge for
compression algorithms: Genetic sequences.
Information Processing & Management, 30(6), 875–
886.
Gulli, A., & Pal, S. (2017). Deep Learning with Keras (p.
318). Packt Publishing Ltd.
Hinton, G. E., & Salakhutdinov, R. R. (2006). Reducing the
dimensionality of data with neural networks. Science,
313(5786), 504–507.
Kamilaris, A., & Prenafeta-Boldú, F. X. (2018). Deep
learning in agriculture: A survey. Computers and
Electronics in Agriculture, 147, 70–90.
Li, Y., Huang, C., Ding, L., Li, Z., Pan, Y., & Gao, X.
(2019). Deep learning in bioinformatics: Introduction,
application, and perspective in the big data era.
Methods, 166, 4–21.
Marchini, J., Howie, B., Myers, S., McVean, G., &
Donnelly, P. (2007). A new multipoint method for
genome-wide association studies by imputation of
genotypes. Nature Genetics,
39(7), 906–913.
Qiu, Y. L., Zheng, H., & Gavaert, O. (2018). A deep
learning framework for imputing missing values in
genomic data. BioRxiv.
Qiu, Y. L., Zheng, H., & Gevaert, O. (2020). Genomic data
imputation with variational auto-encoders.
GigaScience, 9(8).
Rana, S., John, A. H., & Midi, H. (2012). Robust regression
imputation for analyzing missing data. 2012
International Conference on Statistics in Science,
Business and Engineering (ICSSBE) (pp. 1–4).
Presented at the 2012 International Conference on
Statistics in Science, Business and Engineering
(ICSSBE2012), IEEE.
Scheet, P., & Stephens, M. (2006). A fast and flexible
statistical model for large-scale population genotype
data: applications to inferring missing genotypes and
haplotypic phase. American Journal of Human
Genetics, 78(4), 629–644.
Sento, A. (2016). Image compression with auto-encoder
algorithm using deep neural network (DNN). 2016
Management and Innovation Technology International
Conference (MITicon) (p. MIT-99-MIT-103).
Presented at the 2016 Management and Innovation
Technology International Conference (MITicon),
IEEE.
Troyanskaya, O., Cantor, M., Sherlock, G., Brown, P.,
Hastie, T., Tibshirani, R., Botstein, D., et al. (2001).
Missing value estimation methods for DNA
microarrays. Bioinformatics, 17(6), 520–525.
BIOINFORMATICS 2021 - 12th International Conference on Bioinformatics Models, Methods and Algorithms
108

Wang, R., Bai, Y., Chu, Y.-S., Wang, Z., Wang, Y., Sun,
M., Li, J., et al. (2018). DeepDNA: a hybrid
convolutional and recurrent neural network for
compressing human mitochondrial genomes. 2018
IEEE International Conference on Bioinformatics and
Biomedicine (BIBM) (pp. 270–274). Presented at the
2018 IEEE International Conference on Bioinformatics
and Biomedicine (BIBM), IEEE.
Wang, R., Zang, T., & Wang, Y. (2019). Human
mitochondrial genome compression using machine
learning techniques. Human genomics, 13(Suppl 1),
49.International Conference (MITicon) (p. MIT-99-
MIT-103). Presented at the 2016 Management and
Innovation Technology International Conference
(MITicon), IEEE.
Troyanskaya, O., Cantor, M., Sherlock, G., Brown, P.,
Hastie, T., Tibshirani, R., Botstein, D., et al. (2001).
Missing value estimation methods for DNA
microarrays. Bioinformatics, 17(6), 520–525.
Wang, R., Bai, Y., Chu, Y.-S., Wang, Z., Wang, Y., Sun,
M., Li, J., et al. (2018). DeepDNA: a hybrid
convolutional and recurrent neural network for
compressing human mitochondrial genomes. 2018
IEEE International Conference on Bioinformatics and
Biomedicine (BIBM) (pp. 270–274). Presented at the
2018 IEEE International Conference on Bioinformatics
and Biomedicine (BIBM), IEEE.
Wang, R., Zang, T., & Wang, Y. (2019). Human
mitochondrial genome compression using machine
learning techniques. Human genomics, 13(Suppl 1), 49.
A Deep Learning Method to Impute Missing Values and Compress Genome-wide Polymorphism Data in Rice
109
