
Using ASAR for Analysis of Electrogenic and Human Gut
Microbial Communities
Igor Goryanin
1,2 a
, Anatoly Sorokin
2b
and Olga Vasieva
3c
1
Okinawa Institute Science and Technology, Okinawa, Japan
2
University of Edinburgh, Edinburgh, U.K.
3
Ingenet Ltd, U.K.
Keywords: Metagenome Analysis, Pathogens, Human Microbiome, Bioelectrical Systems.
Abstract: In this paper we describe applications of our ASAR package to functional, taxonomic and pathways analysis
of metagenomes and propose future plans and perspectives. To illustrate an analytical potential of ASAR, we
discuss outcomes of several projects. The main focus is made on metabolic plasticity of electrochemically
active microbial communities and a potential role of integrated symbiotic bacterial interactions;
antipathogenic properties of BES, manifested in its capacity to remove some pathogens from waste streams;
and medical applications of this technology. We present ASAR-based metagenome analysis of evolving
bacterial community from distillery waste over period of 36 months in BES environment as an example.
Application of ASAR to personalised analyses of gut microbiome (GM) and the data interpretation based on
publically available association studies are also discussed in this publication.
1 INTRODUCTION
For last years, we have been engaged in development
of Bioelectrochemical Systems (BES)/Microbial Fuel
Cell (MFC) technology for wastewater treatment. The
BES/MFC applies complex interactions between
microbial populations and electrodes to remove
organics and to generate electricity (Gajda et al,
2018). By utilizing biological, chemical, engineering,
and bioinformatics approaches, wee seek to improve
BES/MFC systems for better treatment efficiencies
and electricity generation by understanding and
building almost ideal microbial communities and
developing cost-effective materials.
For better understanding of underlying biological
processes, we have created pipeline for metagenome
analysis. We have developed a new software,
Advanced metagenomic Sequence Analysis in R
(ASAR) (Orakov et al, 2017), which allows
simultaneous analysis and visualization of
taxonomic, functional, and pathways profiles of
bacterial communities from the metagenome data.
We have used ASAR to describe and improve
a
https://orcid.org/0000-0002-8293-774X
b
https://orcid.org/0000-0001-6236-6452
c
https://orcid.org/0000-0002-0047-0606
complex microbial communities and biofilms in
BES/MFC. The ASAR package is available for
researchers worldwide via GitHub. Statistical data
analysis software has been integrated into the ASAR
SA package, which includes capabilities to: plot
Principle Coordinates Analysis (PCoA), perform
distributed stochastic neighbour embedding (t-SNE),
and to retrieve statistics estimated through the use of
the pairwise permutational analysis of variance
(PERMANOVA).
We have also developed flat FBA modelling
software for community metabolomics studies and
integrated it into the ASAR package
(https://github.com/lptolik/asar_fba). Our ASAR DB
(Orakov et al, 2017) includes more than 400
metagenomes and is the largest in the world
electrogenic metagenomes proprietary database.
Via systematic analysis and recording of multiple
samples we also found a range of species within the
anode communities possessing the capacity for
extracellular electron transfer, both via direct contact
and electron shuttles and were able to detect
differential distribution of bacterial groups on the
Goryanin, I., Sorokin, A. and Vasieva, O.
Using ASAR for Analysis of Electrogenic and Human Gut Microbial Communities.
DOI: 10.5220/0009193602530259
In Proceedings of the 13th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2020) - Volume 3: BIOINFORMATICS, pages 253-259
ISBN: 978-989-758-398-8; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
253

carbon cloth and activated carbon granules of the
anode surface (Kiseleva et al, 2015a). We have
successfully applied our tools for identification and
isolation of a new bacterial strain of Thalassospira HJ
(Kiseleva et al, 2015b). Using computational pathway
analysis and metabolic engineering, we have recently
constructed a novel strain of electrogenic bacterium,
Arcobacter butzleri, which allows single analyte
(lactate and acetate) detection and can be used as a
biosensor (Szydlowsky et al, 2020).
Back, in 2015, we have pioneered with taxonomic
and functional analysis of MFC communities from
different geographical location. Taxonomic analysis
showed that Proteobacteria, Bacteroidetes and
Firmicutes were abundant in AD sludge from distinct
climatic zones and constituted the dominant core of
the MFC microbiomes. Functional analysis revealed
species involved in degradation of organic
compounds commonly present in food industry
wastewaters (Kiseleva et al, 2015 a).
Accumulation of methanogenic Archaea was
observed in the electrogenic biofilms, suggesting
competition (Georg et al, 2019, Kaur et al, 2014a,b)
or rather a symbiotic relationship between
electrogenes and methanogens and a possibility for
simultaneous electricity and biogas recovery from
one integrated wastewater treatment system
(Kiseleva et al, 2015a). Using our metagenomic
approach we described the microbial diversity of the
MFCs planktonic and anodic communities derived
from two distinctively different inocula (Kiseleva et
al, 2015a) to illustrate a consistency of the MFC
community structure. Though two different archaea
species, M. barkeri and M. thermautotrophicus,
increased in the bacterial communities of swine and
biogas waste inoculated MFCs, respectively (Vasieva
et al, 2019), presence of Proteobacteria (mostly
Deltaproteobacteria) phylum and eight Geobacter
genus species as the predominant taxa in both MFCs
anodic communities have been demonstrated.
Functional analyses of metagenomes from our lab
scale experiments was sufficient to reveal metabolic
changes between different species of the MFC
dominant genus, Geobacter, suggesting that optimal
nutrient utilization at the lowest electrode potential is
achieved via genome rearrangements and a strong
inter-strain selection, as well as adjustment of the
characteristic syntrophic relationships. These
observations show a certain degree of metabolic and
genomic plasticity of electrochemically active bacteria
and their communities in adaptation to adverse anodic
and cathodic compartments (Szydlowski et al, 2019).
To study a functional adaptation of the
electrogenic bacterial community in more detail we
have constructed a lab scale and enhanced pilot-scale
reactor for nitrate removal in BES. Under applied
potentials, BES biofilms were dominated by
autotrophic denitrifying bacteria with a potential to
accept electrons from the electrode (bacteria genera
of Galionella, Sideroxydans, Thiobacillus) and
heterotrophic bacteria that are capable to accept
electrons from Fe2+ (bacteria genus of Thauera).
Bacterial community analysis based on shotgun
sequencing from the 3-electrode reactors has
confirmed metabolic adaptation of the
electrochemically active bacterial communities to
distinct anodic and cathodic environments.
Functional analyses of metagenomes suggests that
optimal nutrient utilization at the lowest electrode
potential is achieved via genome rearrangements and
a strong inter-strain selection. NADH-quinone
oxidoreductase (nuoB/C/G/L, nuoD. nuoH genes) and
NADH-ubiquinone oxidoreductase (nad3) genes
show the strongest dependence on the applied
potential and their abundancy evolves strongly over
the period of the experiment (Szydlowski et al, 2019).
We have also demonstrated that such evolution
was correlated with functional enrichment in
metagenomes of genes encoding for particular motile
(
motB, flgEF, fgrM) factors and diminishing presence
of genes encoding for virulent factors of several taxa,
especially Enteronacteriacea (Shiegella, Vibrio) and
Firmicutes (Enterococcus, Clostridia, Listeria)
(Vasieva et al, 2019, Ieropoulos et al, 2017).
The projects outlined here aim to demonstrate
capabilities of the ASAR-based approach in
taxonomic and functional analysis of bacterial
communities and detection of the communities’
adaptive processes at the genomic level.
2 EXAMPLE. METAGENOME
ANALYSIS OF DISTILLERY
WASTE MFC
2.1 Experimental Setup
This study was conducted at the Mizuho Shuzo
Awamory Distillery Ltd., in Okinawa, Japan.
Metagenomics changes that occur during the
initiation period of a 60 L serpentine-channelled MFC
treating awamori distillery wastewater at a four-day
retention time (0.54 L h
-1
) at a constant 27˚C in the
laboratory were previously reported (Kiseleva et al,
2015a). Within the first 70 days of operation the MFC
achieved 80% COD removal (2 kg COD m
-3
d
-1
). In
this study we performed a three-year operation of a
BIOINFORMATICS 2020 - 11th International Conference on Bioinformatics Models, Methods and Algorithms
254

multiple-MFC systems deployed at an awamori
facility, operating at a similar flow rate but under
ambient environmental conditions. Chemical analysis
was carried out at the Okinawa Prefecture
Environmental Science Centre.
• Staged installation of 3 tray modules with
serpentine flow, air breathing cathode and
composite activated carbon granule & glassy
carbon cloth anode.
• Each tray module split into two 21L volumes,
total module volume 50L
• Microbial Fuel cell cathode assembly, operated
without catholyte. Pat No: US 8846220 B2
• Modules fed continuously from pilot site storage,
distillery wastewater ratio increased to raise BOD
loading
• pH adjusted manually in dosing tank to pH 6.8 – 7
• Modules underwent retrofitting several times
during operation to increase feed inlets, improve
function
• Nearly 3 years continuous operation
The observed steady increase in power production
over time was consistent with previous reports
showing that MFC power production correlates with
the thickness of the anode-colonizing biofilm (Nevin
et al, 2008). No evidence of longer-term operation
leading to a state of biofilm “exhaustion” in which the
performance of the electrogenic community declines
(Kassongo et al, 2011) were found for this particular
settings.
2.2 Bioinformatics Analysis
Whole genome sequences and 16S sequences were
initially analysed using custom-developed pipeline,
as described elsewhere (Orakov et al, 2017, Menze et
al, 2016), as well as functional analysis using
PALADIN (only applicable to WGS) (Westbroo et al,
2017). To study the selective enrichment of different
samples, PCoA analysis was performed (Anderson,
2001), and plots were generated using EMPeror
online tool. Compositional analysis of the community
was performed in R version 1.4. (Vázquez-Baeza et
al, 2013) with package compositions. Relative
abundance was represented as composition with
absolute geometry (rcomp). One-way ANOVA was
conducted to verify significant difference in
abundances of taxa between reactors (van den
Boogaart et al, 2016). For visualization purposes, five
most abundant genera in the inoculum and five most
abundant genera at the final week sample were
selected. PERMANOVA analysis was performed
with Adonis function from vegan R package
(Oksanen et al, 2018).
Functional (SEED/RAST) (Overbeek et al, 2005),
taxonomic and KEGG Ontology (Kanehisa, 2000)
annotations of reads tagged with md5 IDs from MG-
RAST together with sample metadata, functional and
taxonomic annotation hierarchy trees were generated
and downloaded. Next, functional and taxonomic
annotations were merged by identical md5’s
corresponding to unique read sequences. Then read
counts were summed for reads with same function and
taxon. Functional and taxonomic read annotations to
lowest level were matched to lowest level annotations
in their corresponding hierarchy trees to generate the
whole phylogeny of each read. The result is the 3D
dataset with axes of Functions, Taxonomy and
Metagenome samples with hierarchy for former two.
Our post-annotation analysis and visualization
tool ASAR (Orakov et al, 2017) uses data integration
algorithm to merge taxonomic and functional data
annotated at read level. The resulting 3D dataset with
axes of Functions, Taxonomy and Metagenome
samples is visualized via three heatmaps of each axis
versus two others (F&T, F&M, T&M). Additionally,
KEGG pathway enrichment sorting/heatmap and its
map visualization are implemented. Advantages of
the tool are:
1) Integrated functional and taxonomic analysis;
2) Comparative analysis of pathway enrichments;
3) KEGG pathway maps visualization.
The heatmaps show log abundance of reads
annotated with selected function in particular taxon
within particular community. On the KEGG map
each functional box is split into sections
corresponding to analysed bacterial communities.
Relevant abundancy of each function in each
community is colour coded from green (the lowest) to
dark red (the highest proportion in the community).
2.3 Results
After 18 months of bacterial community evolution
(Fig. 1) sequences associated with ‘Electron donating
reactions’ and ‘Reverse electron flow’ functions have
increased in abundancy only in the anodic
metagenomes.
A following list of functional categories was
associated with sequences which abundance
increased in all MFC chambers (Fig.1):
Arginine-Urea cycle
Heat shock
Riboflavin, FMN, FAD
Fatty acids metabolic cluster
Organic acids
Electron accepting reactions, NAD and NADH
Oxidative stress
Using ASAR for Analysis of Electrogenic and Human Gut Microbial Communities
255
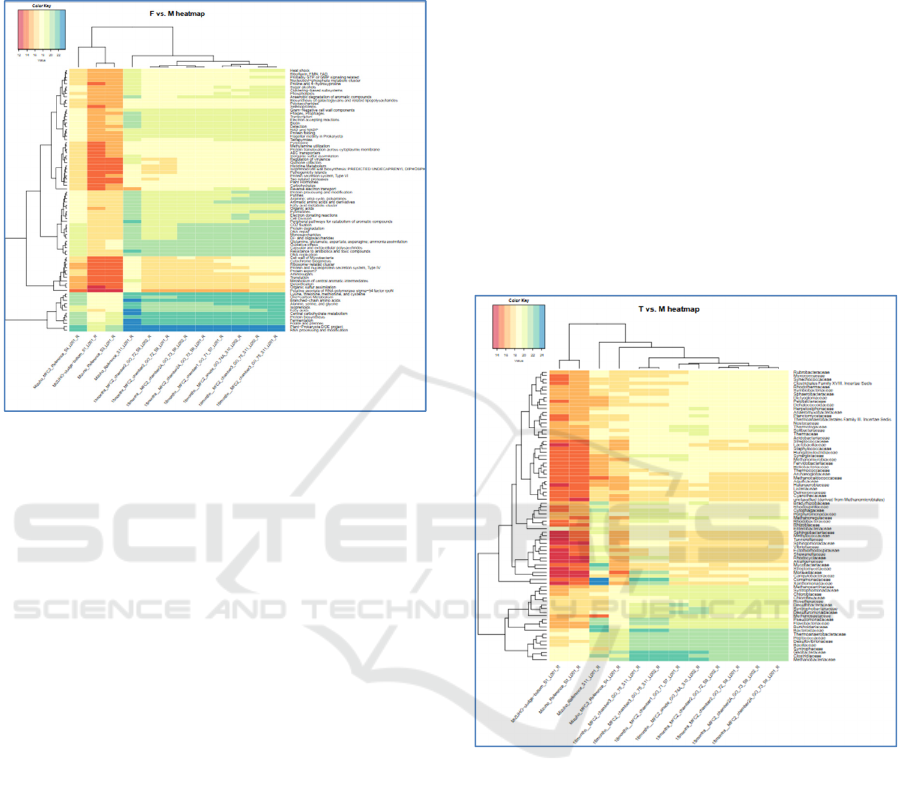
Central carbohydrate metabolism
Fermentation
One carbon metabolism
Figure 1: Relative abundances of reads (log scale) mapped
to bacterial genomes in initial inoculum and in established
MFC communities, generated via the ASAR taxa to
samples function (level 3, max mapper (Orakov et al,
2017)). Log abundance is shown for reads annotated with
selected functions in merged taxa within the metagenomes
from anodic and planktonic communities from different
MFC chambers after 18 months of initial bacterial
community evolution.
After three years of cultivation several bacterial
families became dominating and are specifically
enriched in anodic metagenomes: Geobacteriaceae,
Syntrophoceae, Methanobacteriaceae. We also have
noticed, that families of Clostridiaceae,
Bacteroidoceae, Azonexoceae are increased mainly in
chambers’ metagenomes. At the level of genera the
genomic presentation of following became obviously
abundant in MFC (independently on a particular
MFC’s location): Sytrophobacter, Syntrophus,
Geobacter, Clostridium, Desulfovibrio, Bacteroides,
Methanothermobacter, Thiobacillus, Dechloromonas,
Metahnosphaera, Metahnobrevibacter, Metahnotrix,
Pelobacter, Desulfobacillum. Syntrophobacter,
Syntrophus, Geobacter were stronger presented in
anodic metagenomes. Abundances of genomic
sequence presentation of the following genera were
decreased in anodic metagenomes: Dechloromonas,
Clostridum, Bacteroides, Methanoregula. Here the
difference between the reference (s11) and the MFC
metagenomes became very obvious. (Fig.2)
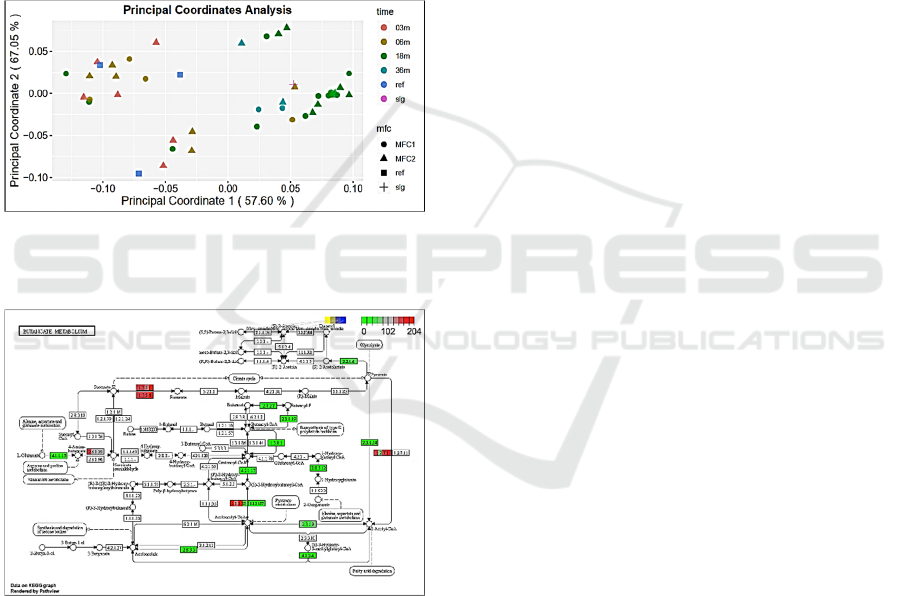
Using Canberra PCoA method (Fig. 3) we
demonstrate a progressive change from the 3 month
community to 18 months community with 36 month
community reversing to positions between 6 and 18
months data points. References data points are close
to 6 months community ones or correspond to the
stage before the 3 month community (close to the
inoculum). Sludge reference data point is placed in
between 18 and 36 months communities’.
Organic acids metabolism and fatty acid
biosynthesis were among the most differentially
expressed between the anodic samples from different
stages of cultivation and in compare to the inoculum
references. It is well presented in the corresponding
KEGG maps. For instance, Fig. 4 presents the KEGG
map for Butanoate metabolism for Geobacteriaceae
family. One can see changes associated with particular
evolutionary time points for 5 key functions.
Figure 2: Relative abundances of reads (log scale) mapped
to bacterial genomes in initial inoculum and in established
MFC communities, generated via the ASAR taxa to
samples function (‘sum’ option mapper (Orakov et al,
2017)). Log abundance is shown for merged reads
annotated for selected taxa within the metagenomes from
anodic and planktonic communities from different MFC
chambers after 18 months of initial bacterial community
(ref) evolution.
3 DISCUSSION
The results of the ASAR-based analysis of the
metagenomes have been implemented in bio-
technological projects that lead to optimisation of the
MFC regimes and the bacterial strains, as well as
generation of new hypothesis, which are awaiting
BIOINFORMATICS 2020 - 11th International Conference on Bioinformatics Models, Methods and Algorithms
256
experimental validations. Our findings pointed to a
potentially antipathogenic property of MFC and
suggested that electrochemical metabolism may be
utilized to supress pathogenic bacteria without
triggering a spread of antibiotics resistance (Vasieva
et al, 2019, Ieropoulos et al, 2017). The highest loss
among pathogenic genera was recorded for
Enterobacteriaceae family (such as Yersinia, Vibrio,
and Shigella). The abundance of virulent genes
responsible for adhesion, secretion systems, invasion,
and intracellular survival, as well as antibiotic
resistance associated with Firmicutes and
Actinobacteria phyla of Gram positive bacteria, also
decreased in the MFC residential metagenomes
(Vasieva et al, 2019).
Figure 3: PCoA analysis for integrated taxonomic and
functional data from metagenomes presenting 2 reference
and 3, 6, 18, 36 months of bacterial community evolution.
Figure 4: Butanoate metabolism. KEGG map for
Geobacteriaceae reads enrichment in anodic communities.
Different colours of parts of each block reflect levels of
abundancies of the corresponding sequences in
metagenomes from different community evolution time
points (as indicated in the inserted legends).
Functional coupling and comparative genomics
analysis have been applied to study functional
associations of Enterococcal cAD1 sex pheromone
precursor (P13268, cad) and its orthologs, known to
be responsible for cell clumping, biofilm formation
and conjugative plasmid transfer associated with
bacterial antibiotics resistance. Our analysis of
genomic neighbourhood, motifs and phylogeny of
cad shows that the cAD1 sex pheromone peptide
release may depend on the precursor’s redox
proprieties, NADH and FMN-based redox
metabolism (NADH oxidoreductase, fumarate
reductase), and a FMN insertion chaperone, flavin
trafficking facilitator ApbE (Q82Z24). We suggest a
hypothetical model linking the NADH-driven and
FMN-dependent redox metabolism and availability
of soluble cofactors with Enterococcus, Listeria,
Oenococcus and the relevant bacterial virulent
properties during the operation of MFC for a purpose
of waste waters and medical waste treatment (Vasieva
and Goryanin, 2019). The novelty of the hypothesised
association between sex pheromone release and the
redox-related enzymatic function of the precursor
lipoprotein suggests a new approach in prevention of
antibiotic resistance spread via targeting sex
pheromone processing chaperones or the cofactor
availability.
We have validated our approach on personalised
gut microbiome (GM) analysis and interpretation
based on published association studies. We have
applied the ASAR to a series of 10 sequenced GMs
from individuals of different ethnical and
geographical backgrounds, age and health groups.
The differentially presented and detectable
taxonomic and functional signatures in each GM
metagenome were used to predict the hosts’
characteristics via correlations established in
published studies, and the predictions were validated
by available individual-associated metadata. We have
tested sensitivity of the routine annotation and data
clustering pipeline to an individual and family-linked
signatures in GM structure and functionalities, when
applied to a limited number of varying samples. The
number of samples was sufficient to demonstrate 2
main types of a GM composition, based on
Bacteroides or Prevotella as the main abundant
genera; limitation of a variety of taxa as a result of
antibiotics application; clustering of family members’
GM metagenomes, both in taxonomic and in
functional space; individual signatures related to
chronic diseases and pharmacological interventions;
and elements of ethnicity related characteristics in the
metagenomes (Vasieva et al, 2019c).
Cross-application of the approach to MFC and
different from MFC’s bacterial communities (such as
Gut microbiomes) (Kaur et al, 2014, Ieropoulos et al,
2017, Vasieva et al, 2019) ensures more detailed
validation of the developed analytical methods, and
sets new standards for their improvement. With more
bacteria genes becoming functionally annotated and
Using ASAR for Analysis of Electrogenic and Human Gut Microbial Communities
257

increasing understanding of metabolic logistics
within an evolved bacterial community we are aiming
to constantly refine our methods. However, it is time
to learn principles of a microbiome adaptive
evolution and the criteria and contrasts that we can
use in the analysis, now and in the future.
4 CONCLUSIONS
We have shown that our computational pipeline and
ASAR package could be successfully used in practical
applications. We have analysed electrogenic and
human microbial communities and produced novel
data used for the software validation and prove of its
capabilities. Original hypothesis were also generated
which require further experimental confirmation.
ACKNOWLEDGEMENTS
We thank OIST support of the research. In particular
OIST Biological Systems Unit members for
providing metagenome sequencing and explanation
of experimental setup.
REFERENCES
Anderson, M. J. 2001. A new method for non-parametric
multivariate analysis of variance. Austral Ecology, 26:
32–46
Gajda, I., Greenman, J., Ieropoulos, I.A., 2018. Recent
advancements in real-world microbial fuel cell
applications. Curr Opin Electrochem, 11:78-83,
doi:10.1016/j.coelec.2018.09.006
Georg, S., de Eguren Cordoba, I., Sleutels. T., Kuntkea, P.,
terHeijneb, A., Buismanab, C. J. N., 2019. Competition
of electrogens with methanogens for hydrogen in
bioanodes. Water Research, 170:115292, doi:
10.1016/j.watres.2019.115292.
Ieropoulos, I., Pasternak, G., Greenman, J., 2017. Urine
disinfection and in situ pathogen killing using a
microbial fuel cell cascade system. PLoS One, 12:1
Kanehisa, M. and Goto, S., 2000. KEGG: Kyoto
Encyclopedia of Genes and Genomes. Nucleic Acids
Res, 28:27-30
Kassongo, J., Togo, C. A., 2011. Performance improvement
of whey-driven microbial fuel cells by acclimation of
indigenous anodophilic microbes. Afr J Biotechnol,
10:7846–7852
Kaur, A., Boghani, H. C., Michie, I., Dinsdale, R. M.,
Guwy, A. J., Premier, G. C., 2014. Inhibition of
methane production in microbial fuel cells: operating
strategies which select electrogens over methanogens.
Bioresource Technology, 173:75-81, doi:
10.1016/j.biortech.2014.09.091
Kiseleva, I., Garushyants, S.K., Ma, H., Simpson, D.J.W.,
Fedorovich, V., Goryanin, I., 2015. Taxonomic and
functional metagenomic analysis of anodic
communities in two pilot-scale microbial fuel cells
treating different industrial wastewaters. J Integr
Bioinform, 12(3):273
Kiseleva, L., Garushyants, S. K., Briliute, J., Simpson,
D.J.W., Cohen, M.F., Goryanin, I., 2015. Genome
sequence of the electrogenic petroleum-degrading
Thalassospira sp. strain HJ Genome Announc. 3 (3),
e00483-15
Menzel, P., Ng, K. L., Krogh, A., 2016. Fast and sensitive
taxonomic classification for metagenomics with Kaiju.
Nature Communications, 7: 11257
Nevin, K. P., Richter, H., Covalla, S. F., Johnson, J. P.,
Woodard, T. L., Orloff, A. L., Jia, H., Zhang, M.,
Lovley, D. R., 2008. Power output and columbic
efficiencies from biofilms of Geobacter sulfurreducens
comparable to mixed community microbial fuel cells.
Environ Microbiol, 10:2505–2514, doi:10.1111/j.1462-
2920.2008.01675.x.
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., 2018.
Vegan: Community Ecology Package. R package, 2:4–6
Orakov, A., Sakenova, N., Goryanin, I., Sorokin A., 2018.
ASAR Database: An R Tool for Visual Analysis and
Storage of Metagenomes in Proceedings of the 11th
International Joint Conference on Biomedical
Engineering Systems and Technologies - Volume 4.
Bioinformatics, 196-200
Orakov, A. N., Sakenova, N. K., Sorokin, A., Goryanin, I.
I., 2017. ASAR: visual analysis of metagenomes in R.
Bioinformatics, 34 (8): 1404-1405
Overbeek, R., Begley, T., Butler, R. M., et al., 2005. The
subsystems approach to genome annotation and its use
in the project to annotate 1000 genomes. Nucleic Acids
Res. 33(17):5691–5702, doi:10.1093/nar/gki866
Szydlowski, L., Sorokin, Vasieva, O., Fedorovich, V.,
Goryanin, I., 2019. Evolutionary dynamics of microbial
communities in bioelectrochemical systems. bioRxiv,
725580
Szydlowsky, L., et al, Goryanin, I., 2020. Novel strain of
Arcobacter isolation and metabolic engineering
(submitted, Metabolic Engineering)
Van den Boogaart, K., Tolosana, R. and Bren, M., 2016.
Compositions: Compositional data analysis r-pack,
http://sp.lyellcollection.org/ Vasieva, O., Sorokin, A.,
Szydlowski, L. and Goryanin, I., 2019. Do Microbial
Fuel Cells have Antipathogenic Properties? J Comput Sci
Syst Biol, 12:3, doi:10.4172/0974-7230.1000301 (a)
Vasieva, O., Goryanin, I., 2019. Is there a function for a sex
pheromone precursor? Journal of Integrative
Bioinformatics, 16(4): 20190016, https://doi.org/10.
1515/jib-2019-0016 (b)
Vasieva, O., Sorokin, A., Murzabaev, M., Babiak, P.,
Goryanin, I., 2019. Study on analysis of personal gut
microbiome. Comput Sci Syst Biol, 12(3):71-79 (.c)
BIOINFORMATICS 2020 - 11th International Conference on Bioinformatics Models, Methods and Algorithms
258

Vázquez-Baeza, Y., M. Pirrung, A., Gonzalez, R., Knight,
2013. EMPeror: a tool for visualizing high-throughput
microbial community data. Gigascience, 2:16
Westbrook, A., Ramsdell, J., Schuelke, T., Normington, L.,
Bergeron, R. D., Thomas, W. K., and MacManes, M.
D., 2017. PALADIN: protein alignment for functional
profiling whole metagenome shotgun data.
Bioinformatics, 33(10):1473–1478
Using ASAR for Analysis of Electrogenic and Human Gut Microbial Communities
259
