
The Behaviour of Neuro-2A Cells on Silicon Substrates with Various
Topographies Generated by Femtosecond Laser Micromachining
Sara Mingu
1* a
, Ihor Pavlov
2b
, Çağdaş Devrim Son
1,3 c
and Alpan Bek
1,2 d
1
Micro and Nanotechnology Graduate Program, Middle East Technical University, 06800, Ankara, Turkey
2
Department of Physics, Middle East Technical University, 06800, Ankara, Turkey
3
Department of Biological Sciences, Middle East Technical University, 06800, Ankara, Turkey
Keywords: Neuro-2A Cells, Ultrafast Laser Processing, Silicon, Live Imaging, Substrate Topography.
Abstract: The interaction of neural cells with silicon surfaces is important for basic research as well as for various
possible applications, such as silicon-based neural implants and neurochips. Laser structuring of silicon
provides a quick and versatile method for the generation of complex, hierarchical topographies on precise
locations of the substrate. The behaviour of Neuro-2A cells with laser-structured silicon substrates was studied
using a live-imaging setup with fluorescence microscopy. Neuro-2A cells were able to adhere to polished
silicon, ripples and microcolumns to different extents, depending on the substrate topography and incubation
time. Initially, cells adhere much better to structured areas, resulting in visible cell patterning on the substrates.
Time-lapse microscopy revealed cell exploration and motility behaviours on the substrates. Cell motility was
significantly decreased on structured substrates, with whole area microcolumns having the slowest cell
motility. On polished silicon, cells were found to interact with the substrates using lamellipodia and filopodia.
After 24 or 48 hours, cells were better able to adhere to polished as well as structured silicon. Neurite
alignment was observed on microcolumn and trench substrates. On the other hand, highly processed substrates
were inhibitory to cell growth and resulted in poor cell health.
1 INTRODUCTION
The interaction of cells with their physical
environment, including substrate topography, is
important in basic and applied research in tissue
engineering and regenerative medicine. Cells have
been shown to respond to substrates via alterations in
adhesion, proliferation, shape or differentiation state
(Dalby, 2014; Yang, 2015).
As a general rule, it is believed that specific parts
of the cells interact with topographical features of
similar size: small sized features (tens of nm) interact
with integrins, micron-sized features interact with
focal adhesions, while larger topographical features
(tens of microns) may interact with the cell bodies. In
particular, neurites were found to interact with
topographical features of various sizes (Yim, 2016).
The underlying biochemical mechanisms have not
been completely elucidated, and there is ongoing
a
https://orcid.org/0000-0002-7332-0764
b
https://orcid.org/0000-0001-9494-4149
c
https://orcid.org/0000-0002-4076-5441
d
https://orcid.org/0000-0002-0190-7945
research on the interaction of cells with substrates.
The parameters to consider are the choice of substrate
materials, the micromachining methods, the substrate
surface topographies, and the cell types.
Different materials such as polymers, glass,
steel, gold, platinum, titanium and silicon have been
shown to be appropriate cell substrate materials, and
various modification techniques have been
characterized (Mendes, 2013). Out of these, silicon
has extensively been studied as a cell substrate, also
due to its importance in microelectronics. It is
possible to generate different topographies on silicon
with lithographic or chemical etching techniques.
Silicon has been proposed as a base material for
implantable electronic devices and has been shown to
be biocompatible in vivo in various forms: bare,
coated with chemicals such as poly-lysine, as well as
in forms of porous silicon, quartz, and fused silica
192
Mingu, S., Pavlov, I., Son, Ç. and Bek, A.
The Behaviour of Neuro-2A Cells on Silicon Substrates with Various Topographies Generated by Femtosecond Laser Micromachining.
DOI: 10.5220/0009160001920199
In Proceedings of the 13th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2020) - Volume 2: BIOIMAGING, pages 192-199
ISBN: 978-989-758-398-8; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

(Khan, 2010). Silicon has been previously studied as
a biomaterial with various cell types.
Direct laser writing is a fast, one-step procedure;
it does not require masks or other treatments, it can
be performed in air, and can be used to structure
silicon (Guo, 2013). Laser processing in silicon
generates complex hierarchical morphologies, which
have features in both micro- and nanoscale and thus
resemble in-vivo topographies. A wide variety of
structures is possible simply by changing the laser
fluence. These include channels/trenches, micro-
columns covered in nano-roughness, as well as laser-
induced periodic surface structures (LIPSS). In
particular, LIPSS may be of two types, high spatial
frequency (HSFL), having a period smaller than the
wavelength of the laser, or low spatial frequency
(LSFL), having a period close to the wavelength. In
our study, LSFL LIPSS is used; to the best of our
knowledge, no other study reports the interaction of
LSFL LIPSS on silicon with cells. Femtosecond-laser
induced LIPSS is interesting because it is a regular
grating-like structure covered in nano-roughness. As
such, it may resemble ordered fibres in the cells’
natural environment. Many studies try to replicate
ordered fibres by polymer electrospinning or
lithographic techniques. However, with laser, unlike
other procedures, selective processing of defined
areas on a single substrate and in a single step is
possible, allowing for simultaneous comparison of
different topographies on the same substrate without
any mask.
Neural cells are particularly important, due to
possible applications for neural prosthetics or chips.
As such, there has been extensive research on the
interaction of various primary neurons or neural stem
cell lines with topographies on different materials
(Simitzi, 2018). In the present study, the Neuro-2A
cell line was chosen, as a fast-growing, widely
available cell line which can differentiate into neural
cells spontaneously or upon induction. Neuro-2A
cells have previously been used in studies of cell-
substrate interactions. In one study, Neuro-2A cell
adhesion and localization was modulated on PDMS
chips subjected to chemical treatments, and
communicating neural networks were formed
(Yaghoub, 2005). In another study, 20 micron-width
grooves on PDMS were found to be optimal for cell
density, neurite alignment and differentiation rate for
Neuro-2A cells (Beduer, 2011). Neuro-2A cells have
also been previously studied on silicon substrates. It
was shown that Neuro-2A cells prefer porous silicon
of pore sizes 8 to 75 nm compared to flat silicon, and
that the cell clustering was changed on the porous
surfaces, possibly forming neural networks (Gentile,
2016).
In this work, Neuro-2A cells were observed
interacting with various topographies on silicon
including ablation based microcolumns and LIPSS
based nanoscale ripples using time-lapse microscopy
and confocal microscopy. To our knowledge, this is
the first study where this cell line is used on laser
structured silicon substrates, including LIPSS
topographies, using live imaging.
2 METHODS
2.1 Laser Structuring of Silicon
Substrates
Single-side polished, p-type silicon substrates were
cut into 0.5 mm or 16 mm side squares using precise
laser cutting, in order to ensure an equal number of
cells seeded onto each substrate. The surfaces were
cleaned with hydrogen fluoride for five minutes
before the laser processing. The Laser Induced
Periodic Surface Structures and other topographies
were created using our home-made ultrafast laser
system with central wavelength at 1030 nm, pulse
duration of 370 fs, repetition rate of 1 MHz, and with
up to 1 W of average power. The focused beam spot
size was 9 μm. The system was equipped with a
waveplate for controlling polarization of the laser
beam on the sample, a 3D motorized stage, as well as
precision motion stages. The laser beam was be
raster-scanned onto the substrate using various
shapes, velocities and hatch values (distance between
raster-scan lines). The laser processing was carried
out in air. On 5 mm square substrates, 3mm square
patterning was done. On the 16 mm square substrates,
12 rectangles of 3 mm x 2.5 mm were structured, of 4
different topographies with 3 replicas, each.
2.2 Substrate Cleaning and
Characterization
Laser-structured silicon substrates were cleaned
using 3 solvents (acetone, absolute ethanol and
isopropanol) for 10 minutes each in an ultrasonic
bath, dried using nitrogen air flow, and stored in a
closed container until characterization or use.
Topographies were characterized using Scanning
Electron Microscopy (Zeiss).
The Behaviour of Neuro-2A Cells on Silicon Substrates with Various Topographies Generated by Femtosecond Laser Micromachining
193

2.3 Cell Culture
Neuroblastoma cells were purchased from ATCC
(Neuro-2A line, albino mouse origin, ATCC® CCL-
131™) and maintained in complete medium
containing Dulbecco’s Modified Eagle Medium
(DMEM) and reduced serum medium (OptiMEM),
supplemented with 10% Fetal Bovine Serum (FBS)
and 1% Penicillin/Streptomycin. For differentiation
purposes, cell medium containing 1% FBS was
prepared. The cells were incubated in 37°C and 5%
CO2 in a Nüve EC 160 incubator. During routine
splitting procedures, the cells were washed using
Phosphate Buffered Saline (PBS) and detached using
TrypLE dissociation reagent. 24 hours prior to cell
seeding, the cells were transiently transfected with
fluorescent protein constructs using Lipofectamine
LTX. The DNA constructs included membrane-
bound or nucleus-localized EGFP constructs
equipped with the appropriate localization signals
(Gap-43 signal for membrane and c-myc NLS for
nucleus, respectively). The DNA constructs were
generated using standard molecular cloning
procedures. The Gap43 signal sequence was
MLCCMRRTKQVEKNDEDQKI, while the NLS
sequence was PAAKRVKLD. The amount of
transfected plasmid was 500 ng in all cases. All cell
culture experiments were carried out in a laminar
flow hood equipped with HEPA filter to ensure sterile
conditions.
2.4 Cell Seeding on the Silicon
Substrates
The silicon substrates were sterilized by immersion in
70% ethanol, and then immersed in complete cell
medium for 10 minutes prior to cell seeding in order
to enhance cell adhesion. The cell number was 4000
cells/cm
2
for long duration experiments and 20.000
cells/cm
2
for shorter duration ones. For live imaging
experiments, the substrates were placed upside down
over confocal glass bottom cell culture dishes. The
imaging was done 24 or 48 hours after seeding for
pictures; and for a duration of 3 to 24 hours after
seeding for live-imaging experiments.
2.5 Cell Imaging and Analysis
The cells were imaged with an inverted fluorescence
microscope (Leica) equipped with a confocal setup
(Andor) and the appropriate excitation and emission
filters for EGFP. The silicon substrates were washed
once with HHBS buffer and placed upside down onto
a coverslip. For live imaging, the dish containing the
silicon sample was placed into a live-imaging
chamber equipped with 37°C incubation and 5% CO
2
pump to ensure proper cell growth conditions. Images
were taken every 2 or 2.5 minutes with 10X objective
to observe cell movement on the substrate. All cell
image analyses were carried out with ImageJ or FIJI
software. Cell motility analysis was carried out using
the Manual Tracking Plugin in ImageJ. At least 7
cells were chosen at random and tracked from time-
lapse videos of cells on flat, LIPSS and microcolumn
topographies. The velocity and distance values were
compared for each topography using t-test.
3 RESULTS AND DISCUSSION
The topographies generated on silicon were
dependent on the scan velocity of the beam (which in
turn controls effective fluence) and the distance
between the raster scan lines (hatch). A velocity of 10
mm/s generated deep trench-like topographies with
depth of more than 1 μm (Figure 1). In the presented
substrate, the distance between the lines is 0.03 mm.
A velocity of 100 mm/s generated microcolumn-like
line topographies, while velocities larger than 1000
mm/s generated regular ripples (in LIPSS regime).
The ripples have depths of 300 nm, periods of 900-
1000 μm, and are covered in nano-scale roughness.
Figure 1: SEM of trench topography generated with a single
laser pass with velocity 10 mm/s, hatch = 0.03 mm, 5.000X
magnification.
Figure 2 shows a combination of the LIPSS with
microcolumn topography, generated by scanning 100
mm/s lines over LIPSS topography. LSFL type
LIPSS topography was generated by raster scanning
over the whole area with velocity 1500 mm/s and
hatch value of 0.005 mm (Figures 2 and 3). The
direction of the ripples depends on the polarization of
BIOIMAGING 2020 - 7th International Conference on Bioimaging
194

the beam. Finally, whole-area microcolumn
topographies were generated by raster-scanning the
beam at 100 mm/s using a hatch value of 0.005 mm.
The resulting topography is shown in Figure 4.
Structured silicon substrates of square shape and 1.6
cm side length are shown in Figure 5. Neuro-2A cells
showed different adhesion and motility on the various
substrates. The cell response was highly dependent on
the initial cell density, presence of clumps, serum
amount in the cell medium, and number of hours of
incubation.
Figure 2: SEM of microcolumn line topography on LIPSS,
generated with velocity 100 mm/s, hatch = 0.03 mm,
overlaid on velocity 2000 mm/s, hatch = 0.005 mm,
10.000X magnification.
Figure 3: SEM of LIPSS topography generated with
velocity 2000 mm/s and hatch = 0.005 mm, 5.000X
magnification.
In general, clumped cells were more likely to
keep adhering to each-other through cell-cell
adhesions and less likely to differentiate when
exposed to low serum media or to respond to the
underlying features. This has previously been
reported for other cell types (Menzies KL., 2010).
Figure 4: SEM of microcolumn topography generated with
velocity 100 mm/s, hatch = 0.005 mm, 10.000X
magnification.
Figure 5: 16 mm square silicon substrates used for
experiments. The three substrates are replicas. Each small
structured area is 3 mm by 2.5 mm. The first row is LIPSS,
as in Figure 3. The second row is microcolumn topography
as in figure 4. The third row is trench topography, as in
Figure 1. The fourth row is microcolumn line topography
on LIPSS, as in figure 2.
One important point is that, although the base
material is silicon, the top part of the substrate is
expected to be a native layer of silicon oxide, SiO
2
, of
depth around 10 nm (Fan, 2002). The substrates were
not treated with any agents such as HF to remove the
top oxide layer. Both Si and SiO
2
were reported to be
non-toxic to cells, but also to not support good cell
adhesion (Fan, 2002).
In the first few hours, cells showed highly
differential adhesion on the polished and structured
substrates. At 3 hours, the cell adhesion was weakest
on the flat regions, with a very low number of cells
staying attached after a single washing step. It is
The Behaviour of Neuro-2A Cells on Silicon Substrates with Various Topographies Generated by Femtosecond Laser Micromachining
195
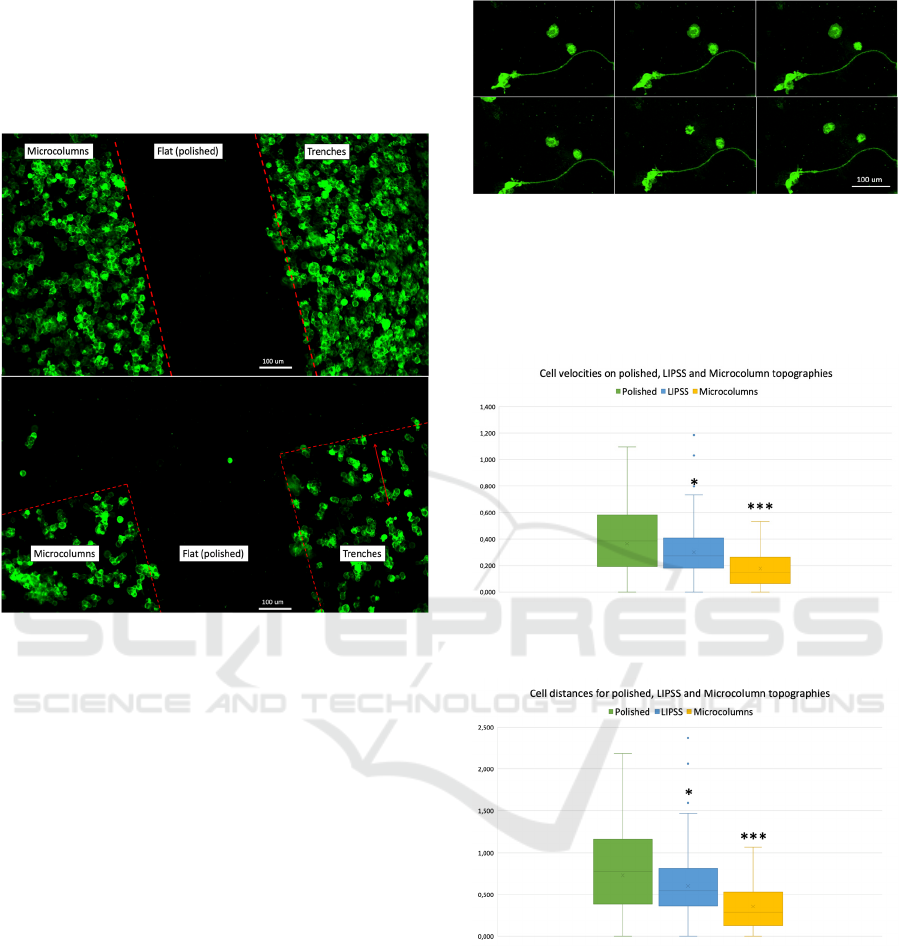
possible that cell adhesion on structured regions
occurs faster, due to the higher surface area of the
structured parts. As a result, cell patterning could be
achieved on the first few hours of incubation. Cell
patterning is shown in Figures 6.
Figure 6: Cell patterning seen on multi-structured substrates
shown in Figure 5, after 3-hour incubation. 20.000
cells/cm
2
were seeded on the substrate. Cells were
expressing membrane localized EGFP. Top and bottom
images are representative edge and vertex regions of the
structured squares and were taken on the same substrate.
Similar cell patterning has been shown for nigral
cells on etched silicon wafers (Fan, 2002) and for
SW10 cells on laser-structured silicon (Yiannakou,
2017). In the latter study, the cells showed a similar
positive response to microcolumn-like topographies,
while being repelled by ripple topographies of very
low periodicity; on the other hand, LSFL LIPSS
regime was not considered. In the present study,
LSFL LIPSS was not found to have any negative
effect on Neuro-2A cell adhesion. The cell patterning
here was exclusively due to adhesion differences,
since cell migration out of the polished regions could
not have occurred at such a short time.
Time-lapse microscopy revealed the cell motility
and exploration patterns on the different structured
surfaces. Time-lapse recordings of cells on polished
silicon, taken 3 hours or 24 hours after seeding
showed exploratory behaviour with lamellipodia and
filopodia. Two representative cells exploring the
polished silicon are shown in Figure 7.
Figure 7: Time-lapse snapshot insets, showing cells moving
on polished silicon. The time-lapse was taken with 10X
objective. The cells had been transfected with membrane
targeted EGFP and had been growing in complete medium
for 16 hours followed by 1% FBS medium for 2 hours. Each
frame corresponds to 6 minutes.
Figure 8: Cell velocities on polished silicon, LIPSS and
microcolumn topographies.
Figure 9: The distances covered by the cells between frames
were significantly different on polished silicon, LIPSS and
microcolumn topographies.
Cell motility, defined as velocity and distance
travelled between frames, was dependent on the
underlying topography. Both velocities and distances
were found to be significantly different among
polished, LIPSS and microcolumn topographies, with
cells moving fastest on polished silicon, significantly
slower on LIPSS (p<0.5, *) and slowest on
microcolumns (p<0.001, ***). Box and whisker plots
of velocities and distances travelled by cells on the
BIOIMAGING 2020 - 7th International Conference on Bioimaging
196
different topographies are shown in Figures 8 and 9,
respectively.
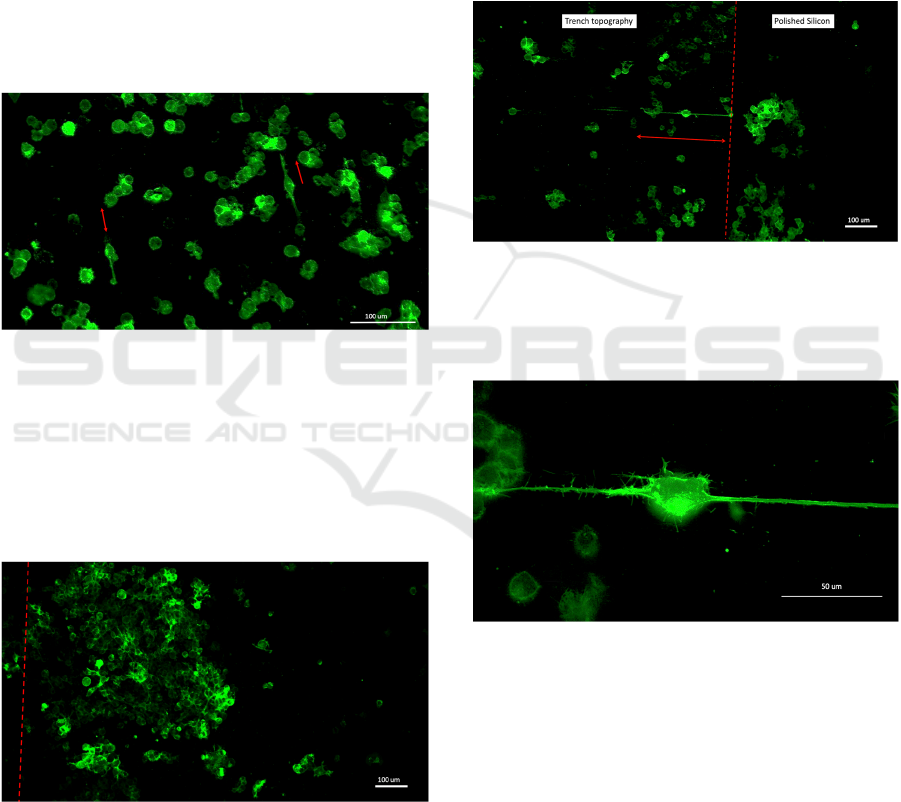
Cells are observed to move preferentially along
the lines back and forth on the topography shown in
Figure 2 and extend one or two neurites along the line
structures reversibly during movement. The width of
the line is around 10 μm (Figure 2), about half the size
of the cell soma. Cells confinement to these lines may
further reflect the cells’ preference to these structures.
However, the migration of these cells is faster than
that of the cells found in whole area microcolumns
(which is evident in live videos), pointing out that it
may be the line topography, and possibly the
interaction of the thin line of the microcolumns with
the underlying LIPSS, which elicits this effect.
Figure 10: Inset of 10X objective time-lapse recording
showing aligned cells on the microcolumn lines on the
topography.
The cell response was different after 24 or 48
hours in culture, possibly due to better adhesion on
polished silicon, or cell movement on the substrates.
More cells remained adhered to polished silicon after
24 or 48 hours, compared to 3 hours.
Figure 11: 10X objective image showing cells on LIPSS,
expressing membrane localized EGFP. The cells had been
growing in complete medium for 24 hours.
When seeded onto line topographies, cells showed
neurite alignment to trenches or microcolumns after
24 or 48 hours in culture. Neurite alignment has
previously been shown in another study, using ridge-
groove substrates generated by multi-step electronic
lithographic methods (Johansson, 2006). In the
present study, a similar effect may be achieved with a
simpler procedure.
On LIPSS, cells were able to remain adhered after
24 or 48 hours. No cell body alignment or elongation
was observed. A representative image of cells
growing on LIPSS is shown in Figure 11. Images of
cells with aligned neurites are shown in Figures 12-
15.
Figure 12: 10X objective image showing various cell
clumps as well as differentiated Neuro-2A cell expressing
membrane localized EGFP and aligned neurites. The
topography consists of trenches, such as in Figure 1. The
cells had been growing in 1% serum medium for 48 hours.
Figure 13: Confocal 63X oil immersion objective image of
the cell shown in Figure 12. The neurites are on the
trenches.
On the other hand, cell body alignment was not
observed for any of the substrates, except for during
cell movement on trenches and microcolumn lines, as
seen in time-lapse videos. Cells which were found
moving on the lines or trenches tended to have a more
elliptical shape compared to the other cells, with the
longer axis coinciding with the topography lines.
However, the shape of the cells was dynamic
throughout the time-lapse imaging.
The cells did not respond well to substrates which
were highly processed. A representative image is
The Behaviour of Neuro-2A Cells on Silicon Substrates with Various Topographies Generated by Femtosecond Laser Micromachining
197
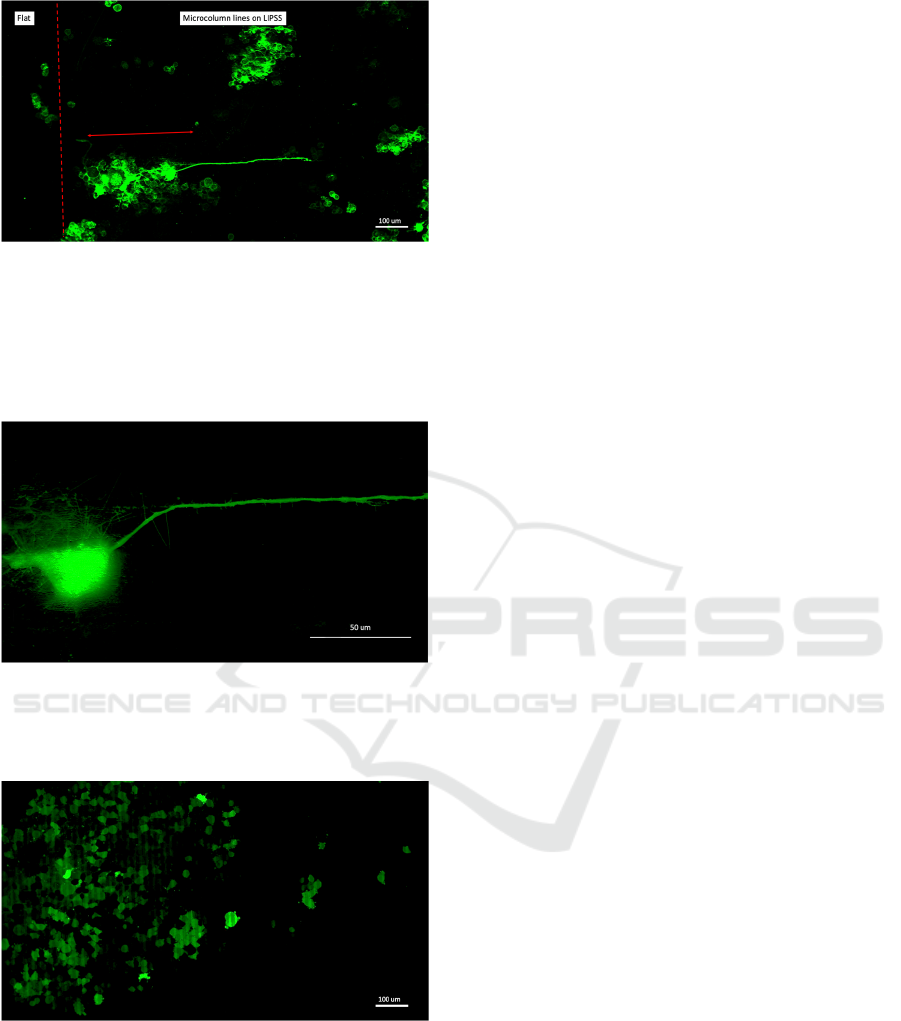
Figure 14: 10X objective image showing various cell
clumps as well as differentiated Neuro-2A cell expressing
membrane-localized EGFP and aligned neurites. The cells
had been growing in 1% serum medium for 24 hours. The
topography consists of microcolumn lines on LIPSS, the
red arrow shows the direction of the microcolumn lines and
LIPSS.
Figure 15: 63X oil immersion objective image of the cell
shown in Figure 14. The neurite extended onto the
microcolumn line. The LIPSS topography can be seen
under the cell body.
Figure 16: 10X magnification image showing blebbing
behaviour of Neuro-2A cells on substrates on highly
processed substrates. The cells had been growing for 24
hours in complete medium and express nuclear-localized
EGFP. The substrate consists of trenches like in Figure 1,
with hatch value of 20 µm.
shown in Figure 16. In these cells, nuclear
localization fluorescent proteins were found
throughout the cell, suggesting that the nuclear
integrity was compromised. Moreover, the cells
display blebbing behaviour. This suggests that it
could be possible to make regions which are cell-
inhibitory in over longer incubation periods, simply
by excessive laser damage.
4 CONCLUSIONS AND FUTURE
WORK
The behaviour of Neuro-2A cells was observed on
different time points on polished and laser-structured
silicon, using confocal fluorescence and time-lapse
microscopy. The cells displayed different behaviours
depending on the topography and incubation time. At
short incubation times, cells were more likely to
attach to the structured areas, opening up the
possibility for easy cell patterning. At later times, the
cell adhesion on all structures was improved, and
neurite alignment effects were observed.
Similar to the cell adhesion, also motility on the
substrates was affected by the underlying
topographies. Further work may include studying the
behaviour of different proteins using live imaging
setups in silicon and other opaque materials, for
example actin dynamics and stress fibre formation on
the different topographies.
Finally, future work may include live observation
of protein-protein interactions on different substrates
with methods such as FRET, which may be valuable
to understand the biochemical mechanisms
underlying the wide variety of cell responses on
topographies.
ACKNOWLEDGEMENTS
Support from TÜBİTAK under grant number
118F375 is kindly acknowledged.
REFERENCES
Beduer, A., Vaysse, L., Flahaut, E., Seichepine, F., Vieu,
C., 2011. Multi-scale engineering for neuronal cell
growth and differentiation. Microelectron. Eng. 88,
1668-1671.
Dalby, M., Gadegaard, N., Oreffo, R., 2014. Harnessing
nanotopography and integrin-matrix interactions to
influence stem cell fate. Nat. Mater. 13, 558-569.
Fan, Y.W., Cui, F.Z., Hou, S.P., Xu, Q.Y., Chen, L.N., Lee,
I.S., 2002. Culture of neural cells on silicon wafers with
nano-scale surface topography. J. Neurosci. Meth. 120,
17-23.
BIOIMAGING 2020 - 7th International Conference on Bioimaging
198

Gentile, F., Marinaro, G., Rocca, R., Berberio, M.,
Cancedda, L., Di Fabrizio, E., Decuzzi, P., 2015.
Networks of neuroblastoma cells on porous silicon
substrates reveal a small world topology. Integr. Biol.
7(2),184–197.
Guo, C., Vorobyev, A., 2013. Direct femtosecond laser
surface nano/microstructuring and its applications.
Laser Photonics Rev. 7(3), 385-407.
Johansson, F., Carlberg, P., Danielsen, N., Montelius, L.,
Kanje, M., 2006. Axonal outgrowth on nano-imprinted
patterns. Biomaterials 27, 1251-1258.
Khan, S., Newaz, G., 2010. A comprehensive review of
surface modification for neural cell adhesion and
patterning. J. Biomed. Mater. Res. 93A(3) 1209-1224.
Mendes, P.M., 2013. Cellular nanotechnology: making
biological interfaces smarter. Chem. Soc. Rev. 42, 9270.
Menzies, K.L., Jones, L., 2010. The impact of contact angle
on the biocompatibility of biomaterials. Optom. Vis.
Sci. 87, 387.
Simitzi, C., Stratakis, E., Karali, K., Ranella, A. 2018.
Controlling the outgrowth and functions of neural stem
cells: the effect of surface topography. ChemPhysChem
19(10), 1143-1163.
Yaghoub, M., Tremblay, R., Voicu, R., Mealing, G.,
Monette, R., Py, C., Faid, K., Sikorska, M. 2005.
Neurogenesis and neuronal communication on
micropatterned neurochips. Biotechnol. Bioeng. 92(3),
336-345.
Yang, L., Liu, H., Yin, Y. 2015. Biomaterial
nanotopography-mediated cell responses: experiment
and modeling. International Journal of Smart and Nano
Materials 5(4), 227-256.
Yiannakou, C., Simitzi, C., Manousaki A., Fotakis C.,
Ranella A., Stratakis E. 2017. Cell patterning via laser
micro/nano structured silicon surfaces. Biofabrication
9, 025024.
Yim, E.N., 2016. From nano to micro: topographical scale
and its impact on cell adhesion, morphology and
contact guidance. J. Phys.: Condens. Matter. 28,
183001.
The Behaviour of Neuro-2A Cells on Silicon Substrates with Various Topographies Generated by Femtosecond Laser Micromachining
199
