
Development of HIV-1 Coreceptor Tropism Classifiers: An Approach
to Improve X4 and R5X4 Viruses Prediction
José Fernando dos Anjos Rodrigues
1a
, Letícia Martins Raposo
1,2 b
and Flavio Fonseca Nobre
1c
1
Programa de Engenharia Biomédica, Universidade Federal do Rio de Janeiro, Av. Horácio Macedo,
2030, Rio de Janeiro, Brazil
2
Departamento de Métodos Quantitativos, Universidade Federal do Estado do Rio de Janeiro, Av. Pasteur,
458, Rio de Janeiro, Brazil
Keywords: Clinical Applications, HIV, Viral Tropism, Genotypic Classifiers.
Abstract: The pathway of human immunodeficiency virus (HIV) infection depends on the composition of a 35-amino
acid variable region in its envelope, known as the V3 loop. Since this discovery, many tools have been
developed to diagnose and predict viral tropism, from biochemical tests to various computational algorithms.
To date, the biggest developmental difficulty is the correct prediction of X4 or R5X4-tropism virions. In this
study, we evaluated some of these recommended criteria and proposed a random forest-based approach for
better prediction of X4-capable (i.e., either X4-only, or R5X4-dual/mixed capability). All methods achieved
a specificity higher than 87%, with geno2pheno 2.5% showing the best performance (98.2%). Nevertheless,
the sensitivity (73.3%) was lower compared to the other approaches. The highest sensitivity was attained by
our Complete Model with an undersampling strategy (90.1%). The accuracy of all approaches ranged from
87.4% to 93.0%. Complete Model with oversampling and Reduced Model with no balancing showed the
highest MCC value (both with 0.796 score). Considering error rates and the number of explanatory variables,
our main objective of increasing the ability to predict viral specimens with X4-tropism was achieved.
1 INTRODUCTION
The Human Immunodeficiency Virus (HIV) is the
etiologic agent of the Acquired Immunodeficiency
Syndrome (AIDS) (Barré-Sinoussi et al., 1983; Gallo
et al., 1983). The virus is known to use the CD4
receptor to infect its host cell as well as a co-receptor,
which might be a CCR5 or CXCR4 receptor
(Clapham & McKnight, 2001).
The management of which coreceptor will be
utilized by HIV in cell infection depends on the
composition of a hyper-variable loop region within
the gp120 protein receptor on the surface of HIV-1
virions called V3 loop. The chemical properties of
amino acids can create an affinity for the chosen
receptor. This affinity is also known as viral tropism
(Schneider-Schaulies, 2000).
HIV tropism is split into three groups: R5
(specimens with CCR5–receptor tropism), X4 (with
a
https://orcid.org/0000-0003-0287-4345
b
https://orcid.org/0000-0003-0613-5582
c
https://orcid.org/0000-0003-4261-8258
CXCR4 tropism), and R5X4 (specimens whose
tropism cannot be determined or have hybrid tropism)
(Berger et al., 1998). Establishing HIV tropism is
crucial to addressing anti-HIV treatment with more
efficient and less harmful strategies. For instance,
Maraviroc is a CCR5-specific inhibitor with mild side
effects (Woollard & Kanmogne, 2015).
Based on this information, many tests have been
developed to determine HIV tropism. The most
accurate is Trofile®, a phenotypical biological
method to identify the tropism of a patient's HIV
(Whitcomb et al., 2007). Unfortunately, this test is
expensive and very time-consuming for clinical
analysis. Still, researchers rely on their findings to
create HIV databases (Poveda et al., 2010).
Other procedures were developed, taking
advantage of genomic sequencing technology and the
increasing use of computational methods in
healthcare. These procedures utilize advanced
Rodrigues, J., Raposo, L. and Nobre, F.
Development of HIV-1 Coreceptor Tropism Classifiers: An Approach to Improve X4 and R5X4 Viruses Prediction.
DOI: 10.5220/0009096101830187
In Proceedings of the 13th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2020) - Volume 3: BIOINFORMATICS, pages 183-187
ISBN: 978-989-758-398-8; ISSN: 2184-4305
Copyright
c
2022 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
183

statistical tools and classification algorithms. Some
genotypic prediction servers are well known, such as
geno2pheno
[coreceptor]
(Lengauer et al., 2007),
WebPSSM (Jensen et al., 2003) and T-CUP (Heider
et al., 2014). However, because of the lack of
available data, these algorithms struggle to predict
X4-capable group correctly. Since most of the
sequences obtained from these viral samples are in
the R5 group, services are better suited to predict
these specimens, resulting in an overfitting issue
(Dietterich, 1995).
In this study, we evaluated some of these
established predictors and proposed a random forest-
based approach to achieve better performing models.
2 MATERIALS AND METHODS
2.1 Data
For this study, we utilized 1622 amino acid sequences
corresponding to the V3 region of HIV-1, subtype B.
1284 samples had R5-tropism and 338 had X4 or
R5X4-tropism (both groups were merged as NR5-
tropism). All sequence information, as well as viral
tropism and subtype, were obtained from the Los
Alamos National Laboratory database
(http://www.hiv.lanl.gov/).
These sequences were converted from the single-
letter amino acid code to numeric representation
using the Engelman’s hydrophobicity scale through
the
peptides R package (Osorio, Rondón-Villarreal,
& Torres, 2015). This scale was used because each
amino acid has a distinct number to represent them
(Engelman, Steitz, & Goldman, 1986).
2.2 Modelling
The sequences were divided into two groups at a ratio
of 70:30, using the
caret package (Kuhn, 2016).
1136 sequences (899 R5 and 237 NR5) were
allocated to the training set and 486 sequences (385
R5 and 101 NR5) to the test group. The same test set
was used to evaluate the models and all the other
predictors involved in this study.
The random forest algorithm (Breiman, 2001) was
employed to build the predictors using the
randomForest R package (Liaw & Wiener, 2002).
To verify if the unbalanced data could influence the
model performance, we also developed models with
oversampling and undersampling strategies.
Oversampling randomly increases the number of
records in the minority class, while undersampling
randomly discards the majority class samples in order
to modify the class distribution.
We also tested if the removal of explanatory
variables with low variance could affect the
performance of the models. Hence, a training set with
all 35 positions of the V3 sequence (henceforth
Complete Model) was used, as well as a training set
without positions with low variability. In this sense,
we used the
nearZeroVar function form caret R
package to remove variables with low variance
(henceforth Reduced Model). Seventeen variables
were removed for the construction of the Reduced
Model. The positions were: 1, 3, 4, 6, 7, 8, 9, 15, 16,
17, 23, 24, 28, 30, 31, 33 and 35. These positions
showed at least 95% conservation among all the
sequences used in this study.
Altogether, six random forest models were
created to predict HIV-1 tropism.
2.3 Genotypic Predictors Comparison
We compared our approach with the following tools:
T-CUP 2.0, geno2pheno
[coreceptor]
and WebPSSM. For
WebPSSM, we used the scoring matrix x4r5, T-CUP
2.0 was used with standard settings and, for
geno2pheno
[coreceptor]
, we used false positive rate
(FPR) cut-off at 2.5%, 5%, and 10%.
2.4 Performance Measures
For the assessment of performance of all tested
algorithms, we calculated the sensitivity, specificity,
accuracy and Matthews Correlation Coefficient
(MCC). The MCC value ‘1’ points to the perfect
prediction, whereas ‘0’ corresponds to a completely
random prediction. The measures are defined as
following:
100
(1)
100
(2)
100
(3)
(4)
as TP stands for the count of true positives, TN true
negatives, FP false positives and FN false negatives.
The NR5 group was stated as the positive class.
The area under receiver operating characteristic
curve (AUC) was also used to evaluate the random
forest model performance. The
pROC R package was
used (Robin et al., 2011). All analyses were
BIOINFORMATICS 2020 - 11th International Conference on Bioinformatics Models, Methods and Algorithms
184
developed in R programming environment, version
3.6.0 (R Development Core Team, 2019).
3 RESULTS
To assess the overall performance of our classifiers,
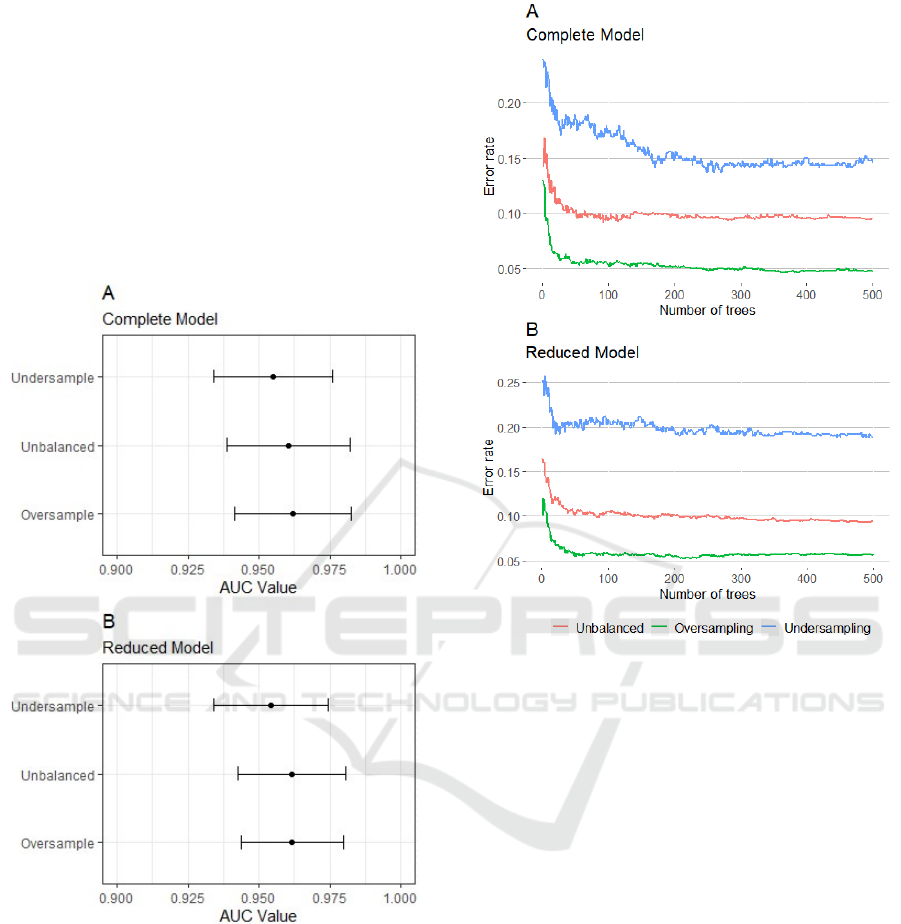
we calculated the AUC. Figure 1 shows the 95%
confidence interval for AUC for each one of the six
models. Both Complete and Reduced models exhibit
very similar performance.
Figure 1: 95% confidence interval for AUC for both
Complete (A) and Reduced (B) models.
The error rate along the construction of decision
trees was also evaluated. In Figure 2, it is possible to
observe that, from 200 trees, the error rates of the
models stabilize. The models built with the
undersampling approach presented higher
fluctuations, while the oversampling model presented
the lowest error rate, both for the Complete and
Reduced models.
We additionally tested our proposed models and
three other methods using an independent test set.
Table 1 shows the performance results of the different
Figure 2: Error rate during the construction of forests for
both Complete (A) and Reduced (B) models.
algorithms. All methods achieved a specificity higher
than 87%, with geno2pheno 2.5% showing the best
performance (98.2%). Nevertheless, the sensitivity
(73.3%) was lower compared to the other approaches.
Our Complete Model with an undersampling strategy
(90.1%) attained the highest sensitivity. The accuracy
of all approaches ranged from 87.4% to 93.0%.
Complete Model with oversampling and Reduced
model with no balancing showed the highest MCC
value (both with 0.796 score).
By comparing the two approaches that use the
random forest algorithms (our models and T-CUP
2.0) it is possible to observe that our classifiers
(Complete with oversampling, Reduced with
oversampling and Reduced with undersampling)
presented a higher performance for the three
evaluated measures.
4 DISCUSSION
In the current study, we developed random forest-
based
approaches for predicting HIV-1 coreceptor
Development of HIV-1 Coreceptor Tropism Classifiers: An Approach to Improve X4 and R5X4 Viruses Prediction
185

Table 1: Performance comparison of different prediction
methods. Highest values for each measure are marked in
bold.
Model Se Sp Acc MCC
Comp Unb 90.1
91.4 91.2 0.759
Comp Over
84.2 94.0 92.0
0.796
Comp Under
88.1 92.5 91.6 0.709
Red Unb
88.1 92.7 91.8
0.796
Red Over
87.1 94.3 92.8 0.753
Red Under
84.2 93.8 91.8 0.722
g2p 10%
86.1 87.8 87.4 0.671
g2p 5%
79.2 93.8 90.7 0.721
g2p 2.5%
73.3
98.2 93.0
0.778
WebPSSM
68.3 93.5 88.3 0.635
T-CUP 2.0
82.2 93.2 90.9 0.733
usage. We used two strategies to enhance our model
performance: methods for balancing training data and
zero or near-zero variance predictors removal. In
total, six approaches were evaluated.
The proposed models performed very similarly to
each other. This information corroborates previous
studies that showed the strength of the random forest
algorithm, even with unbalanced training data
(Dittman, Khoshgoftaar, & Napolitano, 2015).
However, the error rate of the models suggests that
the oversampling approach was more adequate for
this type of problem.
T-CUP 2.0 uses random forest algorithm, like our
model. However, the data preparation for our model
showed influence in its performance. Perhaps, the
choice of Engelman hydrophobicity scale, instead of
Kyte-Doolittle scale (Kyte & Doolittle, 1982), used
for T-CUP 2.0. Therefore, the evaluation of
numerical conversion of amino acids should be
considered as an important factor for the development
of genotypic models.
Regarding the number of explanatory variables in
the model, it was possible to observe that both
approaches (Complete and Reduced models) had
comparable performance, suggesting that there is no
great difference between these models. However, on
behalf of parsimony, it is preferable to have a model
with minimal explanatory variables. Therefore, the
Reduced Model is more suitable to our objective.
The charts also showed that the models barely
change their error rate after 200-250 trees in the
forest, except for the undersampled models. Thus, the
model can perform optimally with a smaller number
of trees, streamlining the process of prediction.
It is very significant that our model has achieved
the highest sensitivity values. Although geno2pheno
algorithm achieved the best performance in
specificity and accuracy, our models showed best
values of MCC, a robust parameter for evaluation of
any prediction method. Our main goal in this study
was to enhance the ability of algorithms to predict
viral specimens with X4 tropism. The Complete
Model with no balancing showed sensitivity and
specificity above 90%, which suit our model into the
European guidelines on the clinical management of
HIV-1 tropism testing (Vandekerckhove et al., 2011).
Therefore, our studies are very promising to achieve
a new and more accurate genotypic predictor.
REFERENCES
Barré-Sinoussi, F., Chermann, J. C., Rey, F., Nugeyre, M.
T., Chamaret, S., Gruest, J., … Montagnier, L. (1983).
Isolation of a T-lymphotropic retrovirus from a patient
at risk for acquired immune deficiency syndrome
(AIDS). Science (New York, N.Y.), 220(4599), 868–
871. https://doi.org/DOI:10.1126/science.6189183
Berger, E. A., Doms, R. W., Fenyö, E.-M. M., Korber, B.
T. M., Littman, D. R., Moore, J. P., … Weiss, R. A.
(1998). A new classification for HIV-1. Nature,
391(6664), 240. https://doi.org/10.1038/34571
Breiman, L. (2001). Random forests. Machine Learning.
https://doi.org/10.1023/A:1010933404324
Clapham, P. R., & McKnight, Á. (2001). HIV-1 receptors
and cell tropism. British Medical Bulletin, 58, 43–59.
https://doi.org/10.1093/bmb/58.1.43
Dietterich, T. (1995). Overfitting and Undercomputing in
Machine Learning. ACM Computing Surveys (CSUR),
27(3), 326–327. https://doi.org/10.1145/212094.212114
Dittman, D. J., Khoshgoftaar, T. M., & Napolitano, A.
(2015). The Effect of Data Sampling When Using
Random Forest on Imbalanced Bioinformatics Data. In
Proceedings - 2015 IEEE 16th International
Conference on Information Reuse and Integration, IRI
2015. https://doi.org/10.1109/IRI.2015.76
Engelman, D. M., Steitz, T. A., & Goldman, A. (1986).
Identifying nonpolar transbilayer helices in amino acid
sequences of membrane proteins. Annual Review of
Biophysics and Biophysical Chemistry.
https://doi.org/10.1146/annurev.bb.15.060186.001541
Gallo, R. C., Sarin, P. S., Gelmann, E. P., Robert-Guroff,
M., Richardson, E., Kalyanaraman, V. S., … Popovic,
M. (1983). Isolation of human T-cell leukemia virus in
acquired immune deficiency syndrome (AIDS).
Science. https://doi.org/10.1126/science.6601823
Heider, D., Dybowski, J. N., Wilms, C., & Hoffmann, D.
(2014). A simple structure-based model for the
prediction of HIV-1 co-receptor tropism. BioData
Mining, 7, 14. https://doi.org/10.1186/1756-0381-7-14
Jensen, M. A., Li, F.-S., van ’t Wout, A. B., Nickle, D. C.,
Shriner, D., He, H.-X., … Mullins, J. I. (2003).
BIOINFORMATICS 2020 - 11th International Conference on Bioinformatics Models, Methods and Algorithms
186

Improved Coreceptor Usage Prediction and Genotypic
Monitoring of R5-to-X4 Transition by Motif Analysis
of Human Immunodeficiency Virus Type 1 env V3
Loop Sequences. Journal of Virology.
https://doi.org/10.1128/jvi.77.24.13376-13388.2003
Kuhn, M. (2016). Package ‘caret.’ Retrieved February 20,
2017, from ftp://cran.r-project.org/pub/R/web/
packages/caret/caret.pdf
Kyte, J., & Doolittle, R. F. (1982). A simple method for
displaying the hydropathic character of a protein.
Journal of Molecular Biology. https://doi.org/10.1016/
0022-2836(82)90515-0
Lengauer, T., Sander, O., Sierra, S., Thielen, A., & Kaiser,
R. (2007). Bioinformatics prediction of HIV coreceptor
usage. Nature Biotechnology, 25(12), 1407–1410.
https://doi.org/10.1038/nbt1371
Liaw, A., & Wiener, M. (2002). Classification and
Regression by randomForest. R News.
Osorio, D., Rondón-Villarreal, P., & Torres, R. (2015).
Peptides: A package for data mining of antimicrobial
peptides. R Journal. https://doi.org/10.32614/rj-2015-
001
Poveda, E., Alcami, J., Paredes, R., Córdoba, J., Gutiérrez,
F., Llibre, J. M., … García, F. (2010). Genotypic
determination of HIV tropism - Clinical and
methodological recommendations to guide the
therapeutic use of CCR5 antagonists. AIDS Reviews.
R Development Core Team. (2019). R Software. R: A
Language and Environment for Statistical Computing.
Robin, X., Turck, N., Hainard, A., Tiberti, N., Lisacek, F.,
Sanchez, J.-C., & Müller, M. (2011). pROC: an open-
source package for R and S+ to analyze and compare
ROC curves. BMC Bioinformatics, 12(1), 77.
https://doi.org/10.1186/1471-2105-12-77
Schneider-Schaulies, J. (2000). Cellular receptors for
viruses: Links to tropism and pathogenesis. Journal of
General Virology. https://doi.org/10.1099/0022-1317-
81-6-1413
Vandekerckhove, L. P. R., Wensing, A. M. J., Kaiser, R.,
Brun-Vézinet, F., Clotet, B., De Luca, A., … Boucher,
C. A. B. (2011). European guidelines on the clinical
management of HIV-1 tropism testing. The Lancet
Infectious Diseases. https://doi.org/10.1016/S1473-
3099(10)70319-4
Whitcomb, J. M., Huang, W., Fransen, S., Limoli, K.,
Toma, J., Wrin, T., … Petropoulos, C. J. (2007).
Development and characterization of a novel single-
cycle recombinant-virus assay to determine human
immunodeficiency virus type 1 coreceptor tropism.
Antimicrobial Agents and Chemotherapy.
https://doi.org/10.1128/AAC.00853-06
Woollard, S. M., & Kanmogne, G. D. (2015). Maraviroc: A
review of its use in hivinfection and beyond. Drug
Design, Development and Therapy.
https://doi.org/10.2147/DDDT.S90580
Development of HIV-1 Coreceptor Tropism Classifiers: An Approach to Improve X4 and R5X4 Viruses Prediction
187
