
Alteration of the Signal Transduction Pathway in RASopathies as a
Basis of Targeted Therapeutic Drug Development
Made Ananda Krisna
1
a
, Yulia Ariani Aswin
2
b
1
Master’s programme in Biomedical Science, Faculty of Medicine, Universitas Indonesia, Jl. Salemba Raya No,6,Central
Jakarta Indonesia
2
Medical Biology Department, Faculty of Medicine, Universitas Indonesia, Jakarta, Indonesia
Keywords: Ras protein, Mitogen-activated protein kinase, ERK, RASopathy, molecular targeted therapy
Abstract: Ras/mitogen-activated protein kinase (MAPK) pathway is one of the most critical intracellular signalling
cascades, relaying the extracellular signal in the form of growth factor into specific responses. The primary
responses of Ras/MAPK pathway activation are cellular proliferation and differentiation. Therefore, a
mutation in genes encoding one of its components or regulators causes a severe developmental disorder.
RASopathy is a group of genetic syndromes originating from a germline mutation in the Ras/MAPK
pathway's regulators' genes encoding components. More than 20 genes are associated with and seven
syndromes included in RASopathy: Noonan, LEOPARD, neurofibromatosis type 1, CM-AVM, Costello,
cardio-facio-cutaneous, and Legius syndrome. Genotype-phenotype associations in RASopathy are
complicated, the mutation in one gene could result in different syndromes, while a mutation in different genes
could cause one syndrome. Molecular diagnostic at the genomic level is crucial in establishing the definitive
diagnosis and as the basis for targeted therapy. Several therapeutic agents target the MAPK pathway, but they
have been mainly utilized in malignancy cases in which aberrant MAPK pathway was detected. Research in
targeted therapeutic drug development in RASopathy is still limited, yet it is eminently needed for further
elaboration.
1 INTRODUCTION
RASopathy is a group of syndromic genetic diseases
due to germline mutation in genes encoding
components or regulators of the Ras/mitogen-
activated protein kinase (MAPK) signalling pathway.
(Rauen, 2013; Romano et al., 2010) This pathway
mediates the effects of growth factors and,
consequently, plays an essential role in the growth of
many cells and tissues. Ras is a GTPase protein
encoded by the RAS gene that works following
growth factor receptors activation, usually in tyrosine
kinase receptor (TKR). The MAPK pathway is one of
the most vital downstream signalling cascades of Ras.
Regulatory nuclear proteins are the most common
final target of this pathway most of which are
transcription factors, histones, and other proteins
having a role in the cell cycle, proliferation,
differentiation, and cellular apoptosis and
a
https://orcid.org/0000-0002-7681-7135
b
https://orcid.org/0000-0002-6640-8530
senescence. (Alberts et al., 2008; Morrison, 2012;
Plotnikov, Zehorai, Procaccia, & Seger, 2011)
Disruptions in the Ras/MAPK pathway will
predictably result in a severe developmental disorder,
either localized or systemic, such as what is found in
RASopathy syndromes. Each type of RASopathy has
distinctive characteristics, although there are still
overlapping pathologies amongst them. Some
common clinical manifestations observed in almost
all RASopathy syndromes are abnormalities in the
craniofacial region; malformation of the heart; skin,
muscles, and ocular findings; neurologic and
cognitive disorder; hypotonia; and a higher risk of
developing malignancy. (Rauen, 2013)
The cumulative incidence of all RASopathy
syndromes is 1 case in every 1000 live births. More
than 20 mutated genes had been identified in
RASopathies, and these genes encode proteins
directly involved in the Ras/MAPK signalling
318
Krisna, M. and Aswin, Y.
Alteration of the Signal Transduction Pathway in RASopathies as a Basis of Targeted Therapeutic Drug Development.
DOI: 10.5220/0010492003180329
In Proceedings of the 1st Jenderal Soedirman International Medical Conference in conjunction with the 5th Annual Scientific Meeting (Temilnas) Consortium of Biomedical Science Indonesia
(JIMC 2020), pages 318-329
ISBN: 978-989-758-499-2
Copyright
c
2021 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved

pathway or their regulators. The relative position of
these proteins in the pathway affects the degree of
clinical severity: the more upstream its involvement
in the pathway, the more severe the phenotypes.
However, this correlation is not clear-cut as clinical
heterogeneity is commonly observed in one specific
RASopathy caused by q disruption in the same gene.
(Rauen, 2013; Tajan, Paccoud, Branka, Edouard, &
Yart, 2018) Several syndromes which belong to the
RASopathy group are 1) Neurofibromatosis type 1
(NF1); 2) Noonan syndrome (NS); 3) NS with
multiple lentigo or LEOPARD syndrome; 4)
Capillary malformation-arteriovenous malformation
(CM-AVM) syndrome; 5) Costello syndrome (CS);
6) Cardio-facio-cutaneous (CFC) syndrome; and 7)
Legius syndrome. These syndromes overlap with
each other in terms of clinical manifestations and
their causal gene. On the other hand, one specific
syndrome could also be caused by a mutation in
different genes. This occurs partly due to the high
degree of cross-linking in the MAPK pathway, and it
has a potential clinical impact, especially for targeted
therapy development. (Rauen, 2013; Wu-Chou et al.,
2018)
As with other syndromic diseases, each
RASopathy type is suspected based on the
constellation of signs and symptoms. However,
etiologic diagnosis to elucidate which gene is
affected, and the type of the mutation affecting it is
paramount. RASopathy syndromes caused by
different genes or different mutation in the same gene
might have distinct hereditary patterns and clinical
courses. This information needs to be addressed in a
genetic counselling session with the patients and their
families. Moreover, to know the exact genetic defect
is the first step in building the precise formula for
personalized medicine through targeted therapy or
even gene therapy. (Bai et al., 2019)
In this review, biomedical aspects of RASopathy,
starting from the Ras/MAPK pathway's role in
healthy to various genetic defects underlying each
syndrome, and how it is related to the latest and future
potential drug development in targeted therapy
RASopathy were assessed.
2 RAS AND MAPK SIGNALING
PATHWAY
The MAPK cascade comprises of four families, all of
which are well-characterized, including classical
MAPK known as ERK1/2, C-Jun N-terminal
kinase/stress-activated protein kinase (JNK/SAPK),
p38 kinase, and ERK 5. (Belov & Mohammadi, 2012;
Plotnikov et al., 2011) In each cascade three main
kinases are sequentially activated, including MAPK
kinase kinase (MAPKKK), MAPK kinase (MAPKK),
and MAP kinase (MAPK). To this date, there are at
least 17 MAPKKK, 8 MAPK, and 10 MAPK
identified in mammalian cells. (Morrison, 2012;
Zhang & Liu, 2002) As kinase is an enzyme catalyzing
phosphorylation reaction, this sequential activation
leads to phosphorylation of the regulatory proteins that
are the signalling pathway's final target. This protein
can be located in different subcellular locations, such
as cytoplasm, mitochondria, Golgi apparatus,
endoplasmic reticulum, and nucleus. However, the
most common and important target protein in the
MAPK signalling cascade is located in the nucleus and
functions as gene expression regulators, whether it is
a transcription factor, transcription activator/
suppressor, or protein modulating chromatin
remodelling. (Plotnikov et al., 2011)
2.1 ERK Pathway
The first and foremost identified MAPK pathway is
the ERK1/2 cascade, and therefore it has been the
benchmark for all other kinase cascades. This cascade
has an important role in transducing extracellular
signals mediated through various receptors,
especially the RTK. Phosphorylation of receptor upon
binding with its ligand provides a docking site for
other proteins. Usually, the docked protein is an
intermediary which promotes binding and subsequent
interaction of other proteins in its vicinity. Such
protein is known as an adaptor, and the most
important adaptor in the MAPK pathway is growth
factor receptor-bound protein 2 (Grb2). The Grb2
protein enables interaction between Ras protein and
its activator, the sevenless (SOS) protein which
functions as a guanine nucleotide exchange factor
(GEF). Ras protein superfamily was named after the
tissue and species from which it was first identified:
Rat Sarcoma factor. It is found in the plasma
membrane's cytoplasmic surface, anchored through a
covalent bond to its lipid moiety. The protein has an
intrinsic GTPase activity which hydrolyzes GTP into
GDP so that the activated form has a very short half-
life. Another protein serving as Ras regulator called
GTPase-activating protein (GAP) promotes Ras
GTPase activity. (Belov & Mohammadi, 2012;
Nandan & Yang, 2011; Zenonos & Kyprianou, 2013)
The activated Ras protein has its GDP dissociated
and prefers binding to GTP. This GTP-binding Ras
can recruit and activate MAPKKK proteins such as
Raf-1 and B-Raf. The exact mechanism for
Alteration of the Signal Transduction Pathway in RASopathies as a Basis of Targeted Therapeutic Drug Development
319

MAPKKK activation has not been defined yet.
However, either dimerization or phosphorylation may
be involved in the process. The MAPKKK protein
phosphorylates MAPKK, the MEK1/2 and it turns
phosphorylates ERK1/2 as the last kinase tier, the
MAPK. The phosphorylated ERK is translocated to
the nucleus and can bind either transcription factor
affecting gene expression or the DNA itself. The
affected genes usually encode for proteins promoting
cellular proliferation, differentiation and survival,
and preventing apoptosis. (Morrison, 2012; Plotnikov
et al., 2011)
2.2 JNK and p38 Pathway
These pathways are functionally different from the
ERK pathway because they operate when intra- or
extracellular stressors are present. Although both JNK
and p38 pathway has its particular protein in each
kinase tier, there is a substantial cross-talk between
them. This cross-talk is kept in check by other
proteins, the scaffold-like JNK-interacting proteins
(JIP) so those specific substrates for each pathway are
concentrated and well-compartmentalized.
Because the JNK pathway is the first to be
known as responding to cellular stress, it is also named
a stress-activated protein kinase (SAPK) pathway.
There are several notable distinctions between the
JNK and classical ERK pathway. First, its MAPKKK
can be activated by proteins having an intrinsic
GTPase activity other than Ras, such as Rac1 and
CDC42. Second, the MAPKKK activation can also be
achieved without those proteins' involvement but
rather directly stimulated by an adaptor (e.g. TRAF).
Third, all proteins at the MAPK level belong to the
JNK protein family, which has a threonine-proline-
tyrosine (TPY) motif in their active domains. The JNK
pathway is mainly detected in cells that respond
rapidly to stress, such as neurons, cells of the immune
system, and cells whose activity under the influence of
insulin. (Morrison, 2012; Plotnikov et al., 2011;
Zhang & Liu, 2002) The p38 pathway has its
particularity: as a MAPK, the p38 protein can undergo
autophosphorylation when it is near other molecules,
for example, certain adaptor protein (Tab1 and ZAP-
70) and an analogue of phosphatidylinositol.
(Morrison, 2012; Plotnikov et al., 2011)/
3 RASOPATHY SYNDROMES
Each syndromic disease classified as RASopathy
results from a defect of at least one gene encoding
signal transduction components in the MAPK
pathway or its regulators. The relationship between
the mutated gene and the resulting clinical syndrome
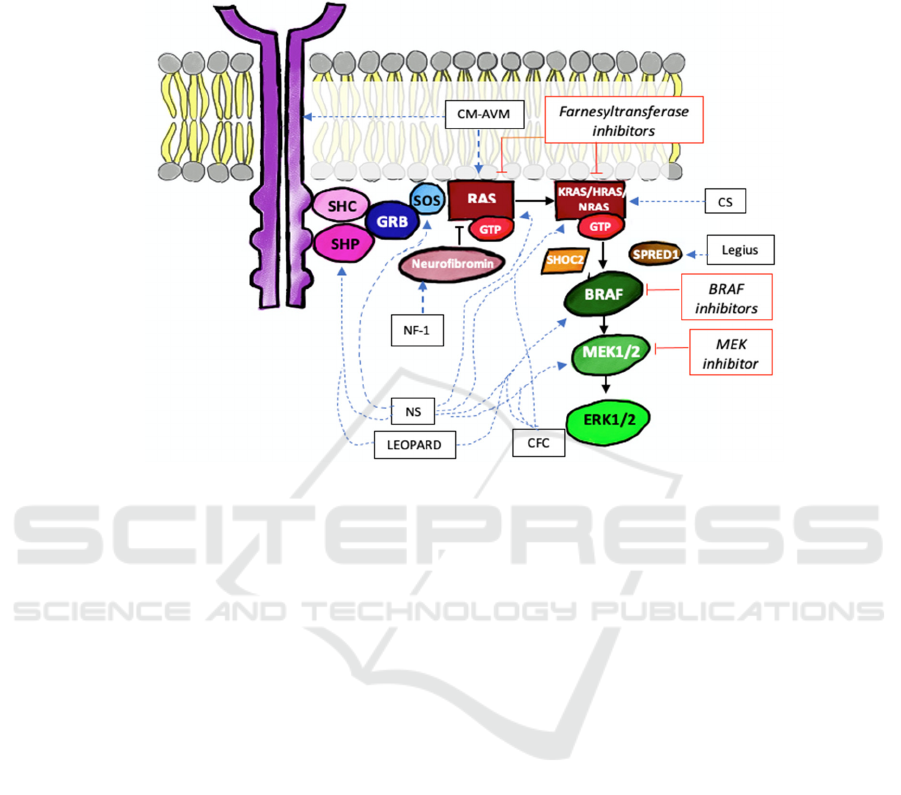
is depicted in Figure 1 as a dashed blue line.
3.1 Noonan Syndrome
Noonan syndrome is a congenital genetic disease with
a relatively high prevalence, the incidence of which is
1 in 1000 to 2500 live births. This syndrome has an
autosomal dominant inheritance pattern with
complete penetrance, but variable expressivity.
Several clinical characteristics can be found in
patients with NS, including the distinctive facial and
musculoskeletal features which include a large skull
with a narrow facial area, wide eyes, prominent
epicanthal folds, ptosis, down-slanting palpebral
fissure, low-set ear, as well as a short nose with a
depressed nasal bridge; thoracic deformity; and
congenital heart disease. (Romano et al., 2010)
Noonan syndrome is a type of RASopathy with
the most heterogeneous genetic defects. The mutated
gene and the type of mutation influence the
phenotype. Mutations in PTPN11, encoding the
enzyme tyrosine phosphatase, consistently show a
significant association with the incidence of thoracic
deformity, mild bleeding disorders, and distinctive
facial features and stature. The nature of the mutation
in that gene also influences the type of heart defect:
missense mutations are associated with a higher
likelihood of pulmonary stenosis and ASD, while a
lower likelihood of cardiomyopathy. Meanwhile,
mutations in SOS1, the gene coding for sons of
sevenless (SOS) protein, have phenotypic
characteristics of the integumentary system that
overlap with CFC syndrome, and mutations in the
SHOC2 gene are associated with different
phenotypes (i.e. mitral valve disorders, growth
hormone deficiency, hyperpigmented skin,
ichthyosis, and developmental disorders). The
Noonan syndrome caused by a KRAS mutation tends
to have a more severe phenotype with more
significant developmental and learning disorders.1
However, not a single phenotype is unique to either a
specific gene or type of mutation. The illustrates the
complexity of interactions between genes in the RAS
/ MAPK pathway. Other than the genes mentioned
above, there are several other causative genes for
Noonan syndrome (Table 1). However, there was no
identified mutation in any genes associated with the
RAS/MAPK pathway in a small number of cases.
(Bai et al., 2019)
JIMC 2020 - 1’s t Jenderal Soedirman International Medical Conference (JIMC) in conjunction with the Annual Scientific Meeting
(Temilnas) Consortium of Biomedical Science Indonesia (KIBI )
320
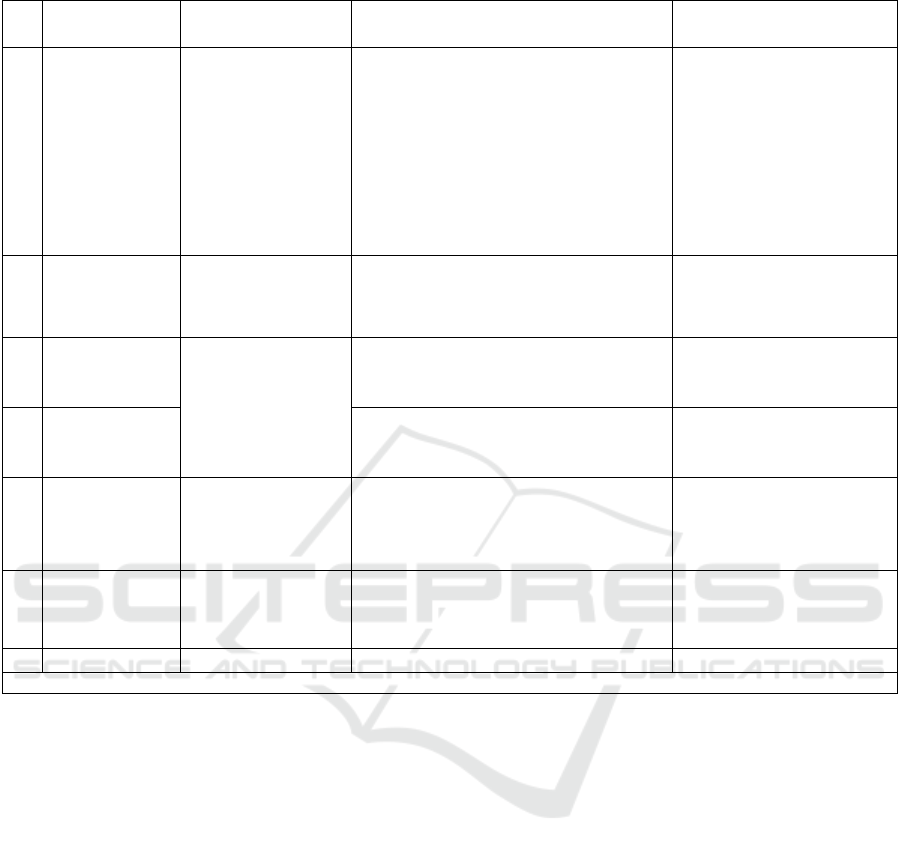
Table 1. Mutated genes and its prevalence in Noonan syndrome.
N
o Gene/protein Subcellular
effects
Characteristics of mutation Prevalence (%)
1 PTPN11/SHP-
2
Cytosolic
phosphatase
cutting
phosphotyrosyl
bond on activated
RTK and
indirectly
activating SOS-
activato
r
Conformational change causing
catalytic site exposure ↑
phosphatase ↑ MAPK activation
42 (Bertola et al., 2006)
58,3 (Chinton et al.,
2019)
60 (F. R. Lepri et al.,
2014)
2 SOS1/Sos As a Ras-
activating GEF
Mutation in autoinhibition sites
↑ GEF activity ↑ RAS/MAPK
pathway activation
11 (Cessans et al., 2016)
18 (F. Lepri et al., 2011)
20 (Roberts et al., 2007)
3 KRAS and
NRAS/Ras
One of Ras
subtypes which
activates
MAPKKK tier
KRAS: ↑ GDP/GTP dissociation
rate ↑ RAS GEF-independent
RAS activation* (Carta et al., 2006)
5
NRAS: de novo somatic
mutation ↑ GAP-resistant RAS
activation (Cirstea et al., 2010)
1 (Cirstea et al., 2010)
4 RIT1/Ras
subfamily
idem ↑ ELK1 activity, a TF which
activates MAPK and activated by the
MAPK pathway(Aoki et al., 2013)
(positive c
y
cle feedback)
3.8
9 (Aoki et al., 2013)
5 RAF1 dan
BRAF/Raf
family
A type of
MAPKKK
RAF1: ↑ Raf activity
BRAF: ↑ Raf activity
15 (Kobayashi et al.,
2010)
1.7
6
M
AP2K1/MEK idem ↑ MEK kinase activit
y
4.2
*Based on structural analysis study digitally (Carta et al., 2006)
3.2 LEOPARD Syndrome
LEOPARD syndrome is also known as Noonan
syndrome with multiple lentigos. The name
"LEOPARD" is an acronym for its main
manifestations: multiple Lentigos,
Electrocardiographic abnormalities, Ocular
hypertelorism, Pulmonary stenosis, Abnormal
genitalia, developmental Retardation, and
sensorineural Deafness. The prevalence of
LEOPARD syndrome in the general population is
unknown. However, it is thought to be significantly
rare because to date, the total number of cases
reported in the publications is no more than 300
patients. (Martinez-Quintana & Rodriguez-Gonzalez,
2012; Sarkozy, Digilio, & Dallapiccola, 2008) This
syndrome is either has an autosomal dominant
inheritance pattern with complete penetrance or
occurred sporadically due to de novo germline
mutation. The main clinical manifestations based on
which the clinical diagnosis is made 1) characteristic
facial features such as hypertelorism, ear
malformations, and low ear with a folded helix; 2)
hypertrophic cardiomyopathy; and 3) café-au-lait
macules which are usually found on the face, neck
and upper part of the torso. Also, several other signs
and symptoms could be encountered in a person with
LEOPARD syndrome, such as thoracic deformity,
cryptorchidism, mild learning, and psychomotor skill
development disorders, and malignancy in the form
of juvenile myelomonocytic leukaemia. (Martinez-
Quintana & Rodriguez-Gonzalez, 2012)
Although the Leopard syndrome is genetically
heterogeneous, 95% of cases have mutations in one
of the following genes: PTPN11, RAF1, and BRAF.
However, as depicted in Table 1, these genes are the
causative genes for more than 50% of NS cases, while
mutations in BRAF are also found in a significant
number of CFC syndrome cases. (Martinez-Quintana
& Rodriguez-Gonzalez, 2012) (Table 2)
Alteration of the Signal Transduction Pathway in RASopathies as a Basis of Targeted Therapeutic Drug Development
321
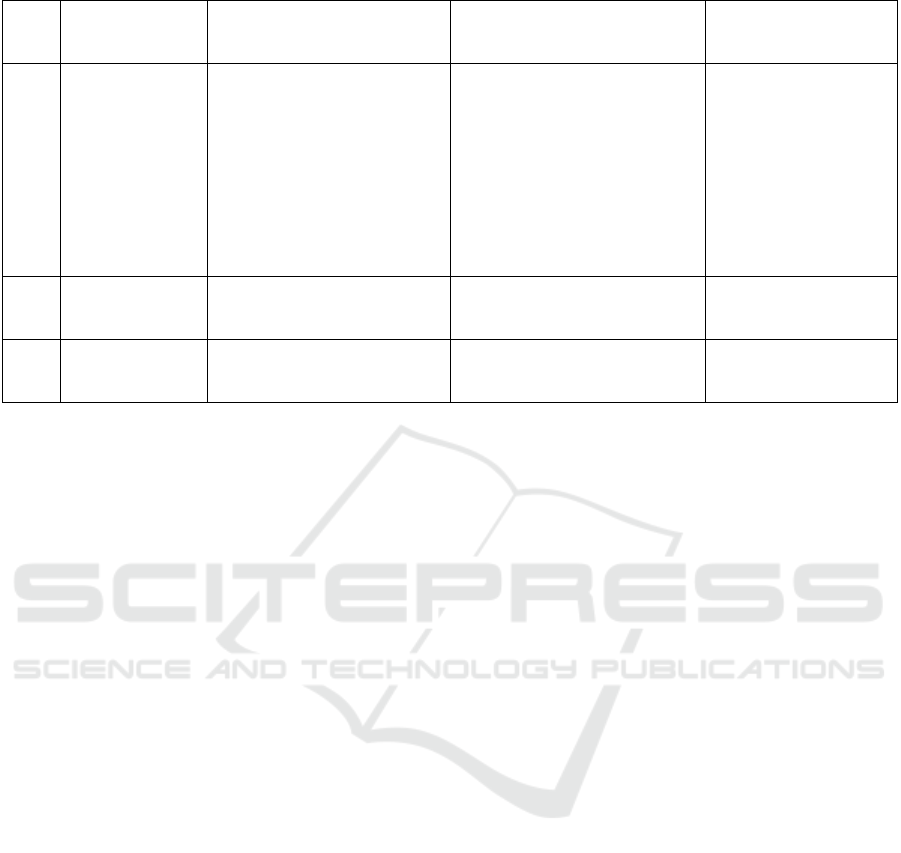
Table 2. Mutated genes and its prevalence in LEOPARD syndrome
No Gene/protein Subcellular effects Characteristics of
mutation
Prevalence (%)
1 PTPN11/SHP-
2
Cytosolic phosphatase
cutting phosphotyrosyl
bond on activated RTK
and indirectly activating
SOS-activator
Mutation inactive catalytic
site ↓ phosphatase but
↑ MAPK activation
42 (Bertola et al.,
2006)
58,3 (Chinton,
Huckstadt, Moresco,
Gravina, & Obregon,
2019)
60 (F. R. Lepri et al.,
2014)
2
RAF1/Raf
family
A type of MAPKKK ↑ Raf activity
<5-10 (Carcavilla et
al., 2013)
3
BRAF/Raf
family
idem Limited ↑ Raf activity
<1-5 (Carcavilla et
al., 2013)
3.3 Neurofibromatosis Type 1
Neurofibromatosis-1 (NF-1) or Von
Recklinghausen's disease is one of the RASopathy
syndromes inherited with an autosomal dominant
inheritance pattern. (Upadhyaya & Cooper, 2012)
The prevalence of NF-1 reaches 1 every 3000 to 4000
individuals in the general population, based on
various studies from Europe and America States.
(Uusitalo et al., 2015) The diagnosis of NF-1 is made
based on clinical manifestations. Some of the most
commonly reported symptoms of NF-1 patients are
café-au-lait macules, neurofibromas of the skin or
oral mucosa, brownish (freckling) spot in the axillary
region, Lisch nodules on the iris, bone abnormalities,
and malignancy, particularly optic nerve gliomas,
astrocytoma, and schwannoma. (Upadhyaya &
Cooper, 2012) The cause of NF-1 is a mutation in the
NF1 gene that encodes the neurofibromin protein.
This protein functions as an activator of GTP-ase
which works on Ras: GTP-ase hydrolyzes GTP bound
to Ras for Ras to reinstate its inactive form. If there is
either a reduction in its level of expression or its
dysfunction, there will be overactivity of the Ras
signalling pathway and subsequent cell growth and
proliferation. (Bennett, Thomas, & Upadhyaya,
2009) Based on previous studies, 100% of patients
with NF-1 have mutations in the NF1 gene even with
heterogeneous types of mutations: it can be small
deletions, missense or nonsense mutations, as well as
splicing mutations. Mutations can accompany
mutations in NF1 in other genes, either the ones
which also play a role in the RAS/MAPK pathway
such as PTPN11 and BRAF, or those which are not,
such as P53. (Arima et al., 2017) Concurrent
mutations in PTPN11 and NF1 genes may indicate a
possibly different clinical entity known as
Neurofibromatosis-Noonan Syndrome (NFNS).
(Thiel et al., 2009; Wu-Chou et al., 2018) Meanwhile,
additional mutations, particularly in other oncogenes
(e.g. P53), increase a patient's probability with NF-1
developing a tumor. (Gottfried, Viskochil, &
Couldwell, 2010)
3.4 Capillary Malformation-arteriovenous
Malformation Syndrome
Capillary malformation-arteriovenous malformation
(CM-AVM) syndrome is a RASopathy syndrome
characterized by multifocal capillary malformations
that are typically found on the face and extremities.
This condition can also be accompanied by
arteriovenous malformations (AVM) and
arteriovenous fistulas. The AVM may occur in
various tissues, including the skin, muscle, bone, and
various internal organs, for instance, the heart and
brain. Consequently, if the AVM ruptures and
bleeding occurs, there will be life-threatening
complications. In general, this syndrome is inherited
with an autosomal dominant pattern. However, in 20-
30% of cases, pathogenic mutations occur de novo.
JIMC 2020 - 1’s t Jenderal Soedirman International Medical Conference (JIMC) in conjunction with the Annual Scientific Meeting
(Temilnas) Consortium of Biomedical Science Indonesia (KIBI )
322
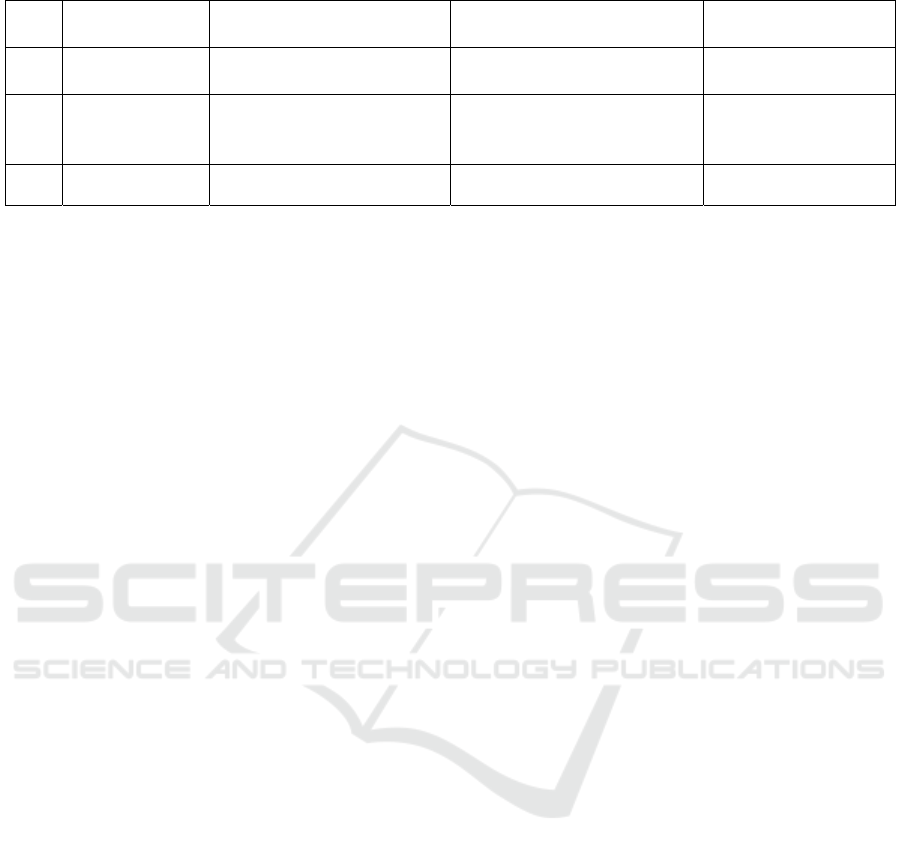
Table 3. Mutated genes and its prevalence in CV-AVM syndrome
No Gene/protein Subcellular effects Characteristics of
mutation
Prevalence (%)
1 RASA1/RASA
1
Ras inactivation ↓ GTPase activity ↑ Ras
activit
y
50 (Amyere et al.,
2017)
2 EPHB4/EphB4 RTK family which signals
through MAPK pathway to
exerts its effects
↑ MAPK pathway activation
10
3 Undetected - - 40
The mutated gene in CM-AVM syndrome can be
either the RASA1 gene or the EPHB4 gene (Table 3).
(Bayrak-Toydemir & Stevenson, 1993-2020) The
RASA1 protein is a negative regulator of Ras through
its activity as a GAP. (Kawasaki et al., 2014)
Meanwhile, the EphB4 protein is a member of the
RTK Eph family which plays a role in embryonic
capillaries morphogenesis. Physiologically, one of
the effects of binding between the EphB4 receptor
and its ligand is the suppression of VEGF-initiated
endothelial proliferation and migration through the
RAS/MAPK signalling cascade. (Kim et al., 2002;
Xiao et al., 2012)
3.5 Costello Syndrome
Costello syndrome's prevalence rate, one of the
RASopathy clinical syndromes, is 1 case per 300,000
live births. This syndrome generally occurs due to
heterogeneous de novo germline mutations in the
HRAS gene. In most cases, the HRAS mutation is
inherited from the father and correlates with an
increase in paternal age. However, in some cases,
somatic HRAS mutations were found. These
mutations lead to continuous activation of the Ras
protein, which causes dysregulation of cell growth
and development. The clinical diagnosis of Costello's
syndrome should be followed by genotyping that
shows the HRAS mutation. If no mutations are found,
the likely diagnosis is not Costello's syndrome, but
other RASopathy syndromes. (Gripp et al., 2019)
Phenotypically, Costello's syndrome has a wide
spectrum of clinical manifestations and may involve
multiple organ systems. In infancy, patients with this
syndrome will experience severe feeding difficulties
that cause growth and developmental problems
including stunting, intellectual disabilities, several
developmental disorders; distinctive facial features
(largemouth, thick lips, large nose tip), papillomas in
the face, and perianal area; generalized hypotonia;
excessive flexibility of the wrist and finger joints; and
involvement of the cardiovascular system in the form
of ventricular hypertrophy, pulmonary stenosis, and
arrhythmias. (Gripp et al., 2019)
3.6 Cardio-Facio-Cutaneous Syndrome
Cardio-facio-cutaneous (CFC) syndrome is
characterized by prominent heart abnormalities
(usually in the form of pulmonary stenosis with other
valves' dysplasia, septal defects, hypertrophic
cardiomyopathy, and arrhythmia), notable
craniofacial characteristics, and concomitant skin
abnormalities including xerosis, hyperkeratosis,
ichthyosis, keratosis pilaris, dermatitis, melanocytic
nevi, hemangioma, and palmoplantar hyperkeratosis.
Almost all patients with CFC syndrome suffered from
neurological and cognitive disorders. Besides, some
of them had significant abnormalities in one or
several other organ systems: musculoskeletal,
lymphatic, ocular, gastrointestinal, and endocrine
systems. Because the clinical manifestations of CFC
syndrome are very diverse, the definitive diagnosis is
established when there is clinical suspicion and
confirmed a pathogenic variant in one of the genes
associated with the syndrome, that is the BRAF,
MAP2K1/2, and KRAS genes. (Table 4) Mutations in
these genes are predominantly de novo and have an
autosomal dominant pattern of inheritance. (Rauen,
1993-2020)
Alteration of the Signal Transduction Pathway in RASopathies as a Basis of Targeted Therapeutic Drug Development
323

Table 4. Mutated genes and its prevalence in CFC syndrome. (Rauen, 1993-2020)
No Gene/protein Subcellular effects Characteristics of mutation Prevalence (%)
1 BRAF/BRAF Activates MAPKK
(MEK1/2)
↑ MAPKK phosphorylation 75
2 MAP2K1/2/M
EK1 dan 2
Activates MAPK
↑ MAPK phosphorylation
25
3 KRAS/KRAS Ras subtype ↓ intrinsic GTPase activity
↑ Ras activit
y
2-3
3.7 Legius Syndrome
The characteristic feature of Legius syndrome is
multiple café au lait macules but without the presence
of neurofibromas or any tumor manifestation as found
in NF-1. Other clinical manifestations that can be
encountered in patients with this syndrome are
intertriginous freckles, lipomas, macrocephaly, and
developmental or learning disorders. To date, only
less than 500 individuals with Legius syndrome have
been reported worldwide with confirmatory
molecular diagnostic laboratory examinations. The
inheritance pattern is autosomal dominant, and
children born to individuals with Legius syndrome
have a 50% chance of inheriting the pathogenic
variant. The only gene associated with the incidence
of this syndrome is the SPRED1 gene. The gene
encodes the Spred1 protein, which physiologically
functions as an inhibitor of the Raf1 kinase activity.
The pathogenic variant of SPRED1 in Legius
syndrome loses its physiological function resulting in
continuous activation of the Raf1 kinase and
increases downstream signalling from the MAPK
pathway. A similar pathophysiological mechanism is
Found in NF-1 with NF1 gene mutation, whose
product's function is very similar to Spred1 protein.
This explains the substantial clinical overlap between
Legius syndrome and NF-1. (Stevenson, Viskochil, &
Mao, 1993)
4 POTENTIAL TARGETED
THERAPEUTIC DRUG
DEVELOPMENT FOR
RASOPATHY
Despite the diverse genetic variations in
RASopathies, the impact of gene mutations in all
syndromes belonging to RASopathy cause an
increase or activation of the RAS/MAPK pathway.
Therefore, targeted therapy to the RAS/MAPK
pathway is predicted to have a role in managing
RASopathy (Figure 1, straight red lines). This
targeted therapy focuses on the downstream part of
the pathway to cover the full spectrum of
pathophysiology in the RASopathy syndromes. Some
of the MAPK pathway blockers include MEK
inhibitors (MAPKK tier), BRAF and Raf inhibitors
(MAPKKK tier), and an antagonist of RAS, the
farnesyltransferase inhibitors. However, the clinical
trials' target population evaluating these drugs was
cancer patients with proven mutations and
dysregulations in the MAPK pathway.
Although some RASopathy syndromes are
associated with an increased risk of developing
malignancy, not all patients with RASopathy exhibit
these clinical manifestations. This has resulted in
excluding most patients with RASopathy syndromes
from many clinical trials evaluating targeted therapy
for the MAPK pathway. (Gross et al., 2020)
Furthermore, most of the studies assessing the use of
inhibitors against components of the MAPK pathway
in RASopathy syndromes were preclinical studies
with the primary objective of explaining disease's
pathophysiological mechanisms, not determining the
effectiveness or efficacy of the inhibitors. However,
all trials reviewed in this article focused on evaluating
the RAS/MAPK pathway inhibitors' effectiveness for
RASopathy, although the majority of which were still
in the preclinical phase.
Ascota et al studied lovastatin therapy in mice
with NF1 mutation, and it was observed that the
attention deficit and spatial aspects of learning were
improved. Lovastatin is a statin drug that inhibits the
HMG-CoA reductase enzyme and acts as an inhibitor
of Ras isoprenylation. This post-translational
modification process is essential for normal Ras
protein function. (Acosta et al., 2011) On the other
hand, Lee et al used a different type of NS animal
model in which the genetic basis of mutation was the
overexpression of the PTPN11 allele. The mice
harboring the mutation increased excitatory synapse
function, deficits in long-term potentiation, and
spatial learning. After the MEK inhibitors
administration, all neurologic manifestations were
alleviated. (Lee et al., 2014) Although not as
prevalent as PTPN11 mutation, SOS1 mutation has
JIMC 2020 - 1’s t Jenderal Soedirman International Medical Conference (JIMC) in conjunction with the Annual Scientific Meeting
(Temilnas) Consortium of Biomedical Science Indonesia (KIBI )
324
also been implicated in NS (Table 1), and an animal
model bearing the mutations were either unviable or
having severe cardiac hypertrophy. Chen et al showed
that prenatal administration of MEK inhibitors to the
mutated mice prevented embryonal lethality and the
cardiac abnormalities that should have occurred
otherwise. (Chen et al., 2010)
Figure 1. RAS/MAPK pathway in RASopathy and potential targeted therapy. Its components and regulators implicated in
RASopathy are shown in elliptical boxes, and the dashed blue lines represent the association between a component/regulator
(if mutated) and each RASopathy syndrome. The red rectangle boxes are potential therapies, and the straight red lines connect
them to their specific targets in RAS/MAPK pathway
Other than NF-1 and NS, there is a relative
scarcity of study on other RASopathy syndromes. A
study by Inoue et al utilized transgenic mice with
BRAF gain-of-function mutation which are most
commonly seen in CFC syndrome, and it showed
consistent phenotypes of embryonic skeletal
abnormalities, lymphatic defects, heart defects, and
liver necrosis. These mutations are lethal in the
embryonic period. However, prenatal administration
of MEK inhibitors causes early embryo development
to return to normal. (Inoue et al., 2014) The Costello
syndrome experimental animal model with gain-of-
Function mutations in HRAS have been shown to
have increased activation of the ERK pathway and
cognitive deficits. In this study, the lovastatin effect
on the syndrome was evaluated. Unlike in the NF1
and NS experimental animal models, its
administration did not restore ERK signalling activity
to baseline level, and the cognitive deficits persisted.
There might have been pathophysiological
differences underlying cognitive deficits among
different syndromes and/or different mutations.
(Schreiber et al., 2017)
Although their numbers are limited, several
studies have demonstrated the pharmacological
potential of MEK inhibitors in human subjects.
Dombi et al. conducted a clinical trial on NF-1
patients with inoperable plexiform neurofibroma
(PN). This benign tumor originates from the myelin
sheath covering the nerves and causes chronic pain,
physical disability, and impaired motor function. The
MEK inhibitor selumetinib administration in this
group of patients significantly reduced tumor size,
reduced pain intensity, and improved motor function.
This study is the first breakthrough to demonstrate the
clinical usefulness of MAPK inhibitors in RASopathy
syndrome and provides preliminary evidence as a
basis for the treatment of other RASopathies (non-
NF-1) using these substances. The United States Food
and Drug Administration has granted permission to
use selumetinib as a PN therapy in NF-1 patients.
(Dombi et al., 2016)
For other RASopathies, as stated before, the
clinical trials were still limited to animal subjects. Wu
et al. used a mice model of NS with mutations in
RAF1 and administered MEK inhibitor PD032590
during the prenatal period. The administration of this
therapy prevented developmental disorders of the
heart. (Wu et al., 2011) Even though it has not been
approved for NS standard therapy, Andelfinger et al
Alteration of the Signal Transduction Pathway in RASopathies as a Basis of Targeted Therapeutic Drug Development
325

applied the MEK inhibitor trametinib to two
newborns with NS who developed heart failure due to
hypertrophic cardiomyopathy. In both of these
infants, myocardial hypertrophy experienced a
significant improvement (partial reversal) within 4
months after the first therapy. (Andelfinger et al.,
2019)
Clinical trials for MAPK-targeted therapy
pathways that have been carried out in the context of
cancer treatment show some substantial side effects
and resistance that results from the initiation of
negative feedback cycles. This can be a problem if
these targeted therapies are used in RASopathy
because the treatment will be continuous and given
for a lifetime. However, the therapeutic doses
required for RASopathy are predictably to be
significantly lower than the doses required to produce
cytotoxic effects in cancer cells. Hence, the likelihood
of side effects is also lower. An essential
consideration for RASopathy targeted therapy is that
most RASopathies result from germline mutations,
while most mutations in cancers are somatic.
Germline mutations underlying RASopathy cause a
homeostatic burden in the form of continuous and
stable tonic activation of the MAPK pathway since
early in life. Consequently, there is a possibility of
harmful regulatory mechanisms in each organ system
as an adaptive attempt to restore homeostasis.
Targeted therapy for the MAPK pathway can be
optimized if supplemented with therapy targeting
these specific adaptation mechanisms. Another
challenge in treating RASopathy using targeted
therapy is that the specific mutation underlying each
syndrome can modify sensitivity to MAPK pathway
inhibitors. For example, several types of mutations in
the PTPN11 gene in NS cause mutant SHP2 protein
production that is resistant to the allosteric inhibitor
of SHP2. Likewise, mutated RAF1 or MEK1 possibly
encode mutant protein resistant to target inhibitor
MEK therapy. (Gripp et al., 2020; Gross et al., 2020;
Tajan et al., 2018)
5 CONCLUSION
RASopathies are a group of syndromic genetic
disorders that result from germline mutations in genes
encoding components or regulators of the Ras /
MAPK signalling pathway. The syndromes included
in RASopathy are Noonan syndrome, LEOPARD
syndrome, neurofibromatosis type 1, CM-AVM
syndrome, Costello syndrome, CFC syndrome, and
Legius syndrome. Clinically, there are plenty of
overlapping signs and symptoms among the
syndromes so that diagnosis based solely on clinical
manifestations is exceptionally challenging. On the
other hand, except for Legius syndrome, genetic
mutations underlying each syndrome are vastly
overlapped to one and another. Therefore, targeted
therapy, especially those that work in the downstream
part of the RAS/MAPK signalling, could be a solution
in the clinical management of RASopathy. All
RASopathy syndromes have in common, both
genotypically and phenotypically, the MAPK
pathway's overactivity. However, treatment in the
form of an inhibitor of the MAPK pathway has been
more widely studied for cases of malignancies with
signalling aberrations in this pathway. Thus, there is
still an opportunity for research development in
specific treatment targeting components of the
MAPK pathway to treat RASopathy syndromes
REFERENCES
Acosta, M. T., Kardel, P. G., Walsh, K. S., Rosenbaum, K.
N., Gioia, G. A., & Packer, R. J. (2011). Lovastatin As
Treatment For Neurocognitive Deficits In
Neurofibromatosis Type 1: Phase I Study. Pediatr
Neurol, 45(4), 241-245.
Doi:10.1016/J.Pediatrneurol.2011.06.016
Alberts, A., Johnson, A., Lewis, J., Morgan, D., Raff, M.,
Roberts, K., & Walter, P. (2008). Molecular Biology Of
The Cell. New York: Garland Science.
Amyere, M., Revencu, N., Helaers, R., Pairet, E., Baselga,
E., Cordisco, M., . . . Vikkula, M. (2017). Germline
Loss-Of-Function Mutations In Ephb4 Cause A Second
Form Of Capillary Malformation-Arteriovenous
Malformation (Cm-Avm2) Deregulating Ras-Mapk
Signaling. Circulation, 136(11), 1037-1048.
Doi:10.1161/Circulationaha.116.026886
Andelfinger, G., Marquis, C., Raboisson, M. J., Theoret, Y.,
Waldmuller, S., Wiegand, G., . . . Hofbeck, M. (2019).
Hypertrophic Cardiomyopathy In Noonan Syndrome
Treated By Mek-Inhibition. J Am Coll Cardiol, 73(17),
2237-2239. Doi:10.1016/J.Jacc.2019.01.066
Aoki, Y., Niihori, T., Banjo, T., Okamoto, N., Mizuno, S.,
Kurosawa, K., . . . Matsubara, Y. (2013). Gain-Of-
Function Mutations In Rit1 Cause Noonan Syndrome,
A Ras/Mapk Pathway Syndrome. Am J Hum Genet,
93(1), 173-180. Doi:10.1016/J.Ajhg.2013.05.021
Arima, Y., Harigai, R., Sato, R., Takenouchi, T., Kosaki,
K., & Saya, H. (2017). Abstract 3418: Additional
Mutation In <Em>Ptpn11</Em> Gene Promotes
Tumorigenesis Of The <Em>Nf1</Em> Gene Mutated
Cells. Cancer Research, 77(13 Supplement), 3418-
3418. Doi:10.1158/1538-7445.Am2017-3418
Bai, R. Y., Esposito, D., Tam, A. J., Mccormick, F.,
Riggins, G. J., Wade Clapp, D., & Staedtke, V. (2019).
Feasibility Of Using Nf1-Grd And Aav For Gene
Replacement Therapy In Nf1-Associated Tumors.
JIMC 2020 - 1’s t Jenderal Soedirman International Medical Conference (JIMC) in conjunction with the Annual Scientific Meeting
(Temilnas) Consortium of Biomedical Science Indonesia (KIBI )
326

Gene Ther, 26(6), 277-286. Doi:10.1038/S41434-019-
0080-9
Bayrak-Toydemir, P., & Stevenson, D. (1993-2020).
Capillary Malformation-Arteriovenous Malformation
Syndrome. In M. P. Adam, H. H. Ardinger, & R. A.
Pagon (Eds.). Retrieved From
Https://Www.Ncbi.Nlm.Nih.Gov/Books/Nbk52764/
Belov, A. A., & Mohammadi, M. (2012). Grb2, A Double-
Edged Sword Of Receptor Tyrosine Kinase Signaling.
Sci Signal, 5(249), Pe49.
Doi:10.1126/Scisignal.2003576
Bennett, E., Thomas, N., & Upadhyaya, M. (2009).
Neurofibromatosis Type 1: Its Association With The
Ras/Mapk Pathway Syndromes. J Pediatr Neurol, 7,
105-115.
Bertola, D. R., Pereira, A. C., Albano, L. M., De Oliveira,
P. S., Kim, C. A., & Krieger, J. E. (2006). Ptpn11 Gene
Analysis In 74 Brazilian Patients With Noonan
Syndrome Or Noonan-Like Phenotype. Genet Test,
10(3), 186-191. Doi:10.1089/Gte.2006.10.186
Carcavilla, A., Santome, J. L., Pinto, I., Sanchez-Pozo, J.,
Guillen-Navarro, E., Martin-Frias, M., . . . Ezquieta, B.
(2013). Leopard Syndrome: A Variant Of Noonan
Syndrome Strongly Associated With Hypertrophic
Cardiomyopathy. Rev Esp Cardiol (Engl Ed), 66(5),
350-356. Doi:10.1016/J.Rec.2012.09.015
Carta, C., Pantaleoni, F., Bocchinfuso, G., Stella, L., Vasta,
I., Sarkozy, A., . . . Tartaglia, M. (2006). Germline
Missense Mutations Affecting Kras Isoform B Are
Associated With A Severe Noonan Syndrome
Phenotype. Am J Hum Genet, 79(1), 129-135.
Doi:10.1086/504394
Cessans, C., Ehlinger, V., Arnaud, C., Yart, A., Capri, Y.,
Barat, P., . . . Edouard, T. (2016). Growth Patterns Of
Patients With Noonan Syndrome: Correlation With
Age And Genotype. Eur J Endocrinol, 174(5), 641-650.
Doi:10.1530/Eje-15-0922
Chen, P. C., Wakimoto, H., Conner, D., Araki, T., Yuan, T.,
Roberts, A., . . . Kucherlapati, R. (2010). Activation Of
Multiple Signaling Pathways Causes Developmental
Defects In Mice With A Noonan Syndrome-Associated
Sos1 Mutation. J Clin Invest, 120(12), 4353-4365.
Doi:10.1172/Jci43910
Chinton, J., Huckstadt, V., Moresco, A., Gravina, L. P., &
Obregon, M. G. (2019). Clinical And Molecular
Characterization Of Children With Noonan Syndrome
And Other Rasopathies In Argentina. Arch Argent
Pediatr, 117(5), 330-337.
Doi:10.5546/Aap.2019.Eng.330
Cirstea, I. C., Kutsche, K., Dvorsky, R., Gremer, L., Carta,
C., Horn, D., . . . Zenker, M. (2010). A Restricted
Spectrum Of Nras Mutations Causes Noonan
Syndrome. Nat Genet, 42(1), 27-29.
Doi:10.1038/Ng.497
Dombi, E., Baldwin, A., Marcus, L. J., Fisher, M. J., Weiss,
B., Kim, A., . . . Widemann, B. C. (2016). Activity Of
Selumetinib In Neurofibromatosis Type 1-Related
Plexiform Neurofibromas. N Engl J Med, 375(26),
2550-2560. Doi:10.1056/Nejmoa1605943
Gottfried, O. N., Viskochil, D. H., & Couldwell, W. T.
(2010). Neurofibromatosis Type 1 And Tumorigenesis:
Molecular Mechanisms And Therapeutic Implications.
Neurosurg Focus, 28(1), E8.
Doi:10.3171/2009.11.Focus09221
Gripp, K. W., Morse, L. A., Axelrad, M., Chatfield, K. C.,
Chidekel, A., Dobyns, W., . . . Rauen, K. A. (2019).
Costello Syndrome: Clinical Phenotype, Genotype,
And Management Guidelines. Am J Med Genet A,
179(9), 1725-1744. Doi:10.1002/Ajmg.A.61270
Gripp, K. W., Schill, L., Schoyer, L., Stronach, B., Bennett,
A. M., Blaser, S., . . . Ratner, N. (2020). The Sixth
International Rasopathies Symposium: Precision
Medicine-From Promise To Practice. Am J Med Genet
A, 182(3), 597-606. Doi:10.1002/Ajmg.A.61434
Gross, A. M., Frone, M., Gripp, K. W., Gelb, B. D.,
Schoyer, L., Schill, L., . . . Yohe, M. E. (2020).
Advancing Ras/Rasopathy Therapies: An Nci-
Sponsored Intramural And Extramural Collaboration
For The Study Of Rasopathies. Am J Med Genet A,
866-876. Doi:10.1002/Ajmg.A.61485
Inoue, S., Moriya, M., Watanabe, Y., Miyagawa-Tomita,
S., Niihori, T., Oba, D., . . . Aoki, Y. (2014). New Braf
Knockin Mice Provide A Pathogenetic Mechanism Of
Developmental Defects And A Therapeutic Approach
In Cardio-Facio-Cutaneous Syndrome. Hum Mol
Genet, 23(24), 6553-6566. Doi:10.1093/Hmg/Ddu376
Kawasaki, J., Aegerter, S., Fevurly, R. D., Mammoto, A.,
Mammoto, T., Sahin, M., . . . Chan, J. (2014). Rasa1
Functions In Ephb4 Signaling Pathway To Suppress
Endothelial Mtorc1 Activity. J Clin Invest, 124(6),
2774-2784. Doi:10.1172/Jci67084
Kim, I., Ryu, Y. S., Kwak, H. J., Ahn, S. Y., Oh, J. L.,
Yancopoulos, G. D., . . . Koh, G. Y. (2002). Ephb
Ligand, Ephrinb2, Suppresses The Vegf- And
Angiopoietin 1-Induced Ras/Mitogen-Activated
Protein Kinase Pathway In Venous Endothelial Cells.
Faseb J, 16(9), 1126-1128. Doi:10.1096/Fj.01-0805fje
Kobayashi, T., Aoki, Y., Niihori, T., Cave, H., Verloes, A.,
Okamoto, N., . . . Matsubara, Y. (2010). Molecular And
Clinical Analysis Of Raf1 In Noonan Syndrome And
Related Disorders: Dephosphorylation Of Serine 259
As The Essential Mechanism For Mutant Activation.
Hum Mutat, 31(3), 284-294. Doi:10.1002/Humu.21187
Lee, Y. S., Ehninger, D., Zhou, M., Oh, J. Y., Kang, M.,
Kwak, C., . . . Silva, A. J. (2014). Mechanism And
Treatment For Learning And Memory Deficits In
Mouse Models Of Noonan Syndrome. Nat Neurosci,
17(12), 1736-1743. Doi:10.1038/Nn.3863
Lepri, F., De Luca, A., Stella, L., Rossi, C., Baldassarre, G.,
Pantaleoni, F., . . . Tartaglia, M. (2011). Sos1 Mutations
In Noonan Syndrome: Molecular Spectrum, Structural
Insights On Pathogenic Effects, And Genotype-
Phenotype Correlations. Hum Mutat, 32(7), 760-772.
Doi:10.1002/Humu.21492
Lepri, F. R., Scavelli, R., Digilio, M. C., Gnazzo, M.,
Grotta, S., Dentici, M. L., . . . Dallapiccola, B. (2014).
Diagnosis Of Noonan Syndrome And Related
Disorders Using Target Next Generation Sequencing.
Alteration of the Signal Transduction Pathway in RASopathies as a Basis of Targeted Therapeutic Drug Development
327

Bmc Med Genet, 15, 14. Doi:10.1186/1471-2350-15-
14
Martinez-Quintana, E., & Rodriguez-Gonzalez, F. (2012).
Leopard Syndrome: Clinical Features And Gene
Mutations. Mol Syndromol, 3(4), 145-157.
Doi:10.1159/000342251
Morrison, D. K. (2012). Map Kinase Pathways. Cold
Spring Harb Perspect Biol, 4(11), A011254.
Doi:10.1101/Cshperspect.A011254
Nandan, M. O., & Yang, V. W. (2011). An Update On The
Biology Of Ras/Raf Mutations In Colorectal Cancer.
Curr Colorectal Cancer Rep, 7(2), 113-120.
Doi:10.1007/S11888-011-0086-1
Plotnikov, A., Zehorai, E., Procaccia, S., & Seger, R.
(2011). The Mapk Cascades: Signaling Components,
Nuclear Roles And Mechanisms Of Nuclear
Translocation. Biochim Biophys Acta, 1813(9), 1619-
1633. Doi:10.1016/J.Bbamcr.2010.12.012
Rauen, K. A. (1993-2020). Cardiofaciocutaneous
Syndrome [Internet]. In M. P. Adam, H. H. Ardinger,
& R. A. Pagon (Eds.). Retrieved From
Https://Www.Ncbi.Nlm.Nih.Gov/Books/Nbk1186/
Rauen, K. A. (2013). The Rasopathies. Annu Rev
Genomics Hum Genet, 14, 355-369.
Doi:10.1146/Annurev-Genom-091212-153523
Roberts, A. E., Araki, T., Swanson, K. D., Montgomery, K.
T., Schiripo, T. A., Joshi, V. A., . . . Kucherlapati, R. S.
(2007). Germline Gain-Of-Function Mutations In Sos1
Cause Noonan Syndrome. Nat Genet, 39(1), 70-74.
Doi:10.1038/Ng1926
Romano, A. A., Allanson, J. E., Dahlgren, J., Gelb, B. D.,
Hall, B., Pierpont, M. E., . . . Noonan, J. A. (2010).
Noonan Syndrome: Clinical Features, Diagnosis, And
Management Guidelines. Pediatrics, 126(4), 746-759.
Doi:10.1542/Peds.2009-3207
Sarkozy, A., Digilio, M. C., & Dallapiccola, B. (2008).
Leopard Syndrome. Orphanet J Rare Dis, 3, 13.
Doi:10.1186/1750-1172-3-13
Schreiber, J., Grimbergen, L. A., Overwater, I., Vaart, T.
V., Stedehouder, J., Schuhmacher, A. J., . . . Elgersma,
Y. (2017). Mechanisms Underlying Cognitive Deficits
In A Mouse Model For Costello Syndrome Are Distinct
From Other Rasopathy Mouse Models. Sci Rep, 7(1),
1256. Doi:10.1038/S41598-017-01218-0
Stevenson, D., Viskochil, D., & Mao, R. (1993). Legius
Syndrome. In M. P. Adam, H. H. Ardinger, R. A.
Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, & A.
Amemiya (Eds.), Genereviews((R)). Seattle (Wa).
Tajan, M., Paccoud, R., Branka, S., Edouard, T., & Yart, A.
(2018). The Rasopathy Family: Consequences Of
Germline Activation Of The Ras/Mapk Pathway.
Endocr Rev, 39(5), 676-700. Doi:10.1210/Er.2017-
00232
Thiel, C., Wilken, M., Zenker, M., Sticht, H., Fahsold, R.,
Gusek-Schneider, G. C., & Rauch, A. (2009).
Independent Nf1 And Ptpn11 Mutations In A Family
With Neurofibromatosis-Noonan Syndrome. Am J Med
Genet A, 149a(6), 1263-1267.
Doi:10.1002/Ajmg.A.32837
Upadhyaya, M., & Cooper, D. N. (2012).
Neurofibromatosis Type 1: Molecular And Cellular
Biology. Berlin: Springer-Verlag Berlin Heidelberg.
Uusitalo, E., Leppavirta, J., Koffert, A., Suominen, S.,
Vahtera, J., Vahlberg, T., . . . Peltonen, S. (2015).
Incidence And Mortality Of Neurofibromatosis: A
Total Population Study In Finland. J Invest Dermatol,
135(3), 904-906. Doi:10.1038/Jid.2014.465
Wu, X., Simpson, J., Hong, J. H., Kim, K. H., Thavarajah,
N. K., Backx, P. H., . . . Araki, T. (2011). Mek-Erk
Pathway Modulation Ameliorates Disease Phenotypes
In A Mouse Model Of Noonan Syndrome Associated
With The Raf1(L613v) Mutation. J Clin Invest, 121(3),
1009-1025. Doi:10.1172/Jci44929
Wu-Chou, Y. H., Hung, T. C., Lin, Y. T., Cheng, H. W.,
Lin, J. L., Lin, C. H., . . . Chen, Y. R. (2018). Genetic
Diagnosis Of Neurofibromatosis Type 1: Targeted
Next- Generation Sequencing With Multiple Ligation-
Dependent Probe Amplification Analysis. J Biomed
Sci, 25(1), 72. Doi:10.1186/S12929-018-0474-9
Xiao, Z., Carrasco, R., Kinneer, K., Sabol, D., Jallal, B.,
Coats, S., & Tice, D. A. (2012). Ephb4 Promotes Or
Suppresses Ras/Mek/Erk Pathway In A Context-
Dependent Manner: Implications For Ephb4 As A
Cancer Target. Cancer Biol Ther, 13(8), 630-637.
Doi:10.4161/Cbt.20080
Zenonos, K., & Kyprianou, K. (2013). Ras Signaling
Pathways, Mutations And Their Role In Colorectal
Cancer. World J Gastrointest Oncol, 5(5), 97-101.
Doi:10.4251/Wjgo.V5.I5.97
Zhang, W., & Liu, H. T. (2002). Mapk Signal Pathways In
The Regulation Of Cell Proliferation In Mammalian
Cells. Cell Res, 12(1), 9-18.
Doi:10.1038/Sj.Cr.7290105
Amyere, M., Revencu, N., Helaers, R., Pairet, E., Baselga,
E., Cordisco, M., . . . Vikkula, M. (2017). Germline
Loss-Of-Function Mutations In Ephb4 Cause A Second
Form Of Capillary Malformation-Arteriovenous
Malformation (Cm-Avm2) Deregulating Ras-Mapk
Signaling. Circulation, 136(11), 1037-1048.
Doi:10.1161/Circulationaha.116.026886
Aoki, Y., Niihori, T., Banjo, T., Okamoto, N., Mizuno, S.,
Kurosawa, K., . . . Matsubara, Y. (2013). Gain-Of-
Function Mutations In Rit1 Cause Noonan Syndrome,
A Ras/Mapk Pathway Syndrome. Am J Hum Genet,
93(1), 173-180. Doi:10.1016/J.Ajhg.2013.05.021
Bertola, D. R., Pereira, A. C., Albano, L. M., De Oliveira,
P. S., Kim, C. A., & Krieger, J. E. (2006). Ptpn11 Gene
Analysis In 74 Brazilian Patients With Noonan
Syndrome Or Noonan-Like Phenotype. Genet Test,
10(3), 186-191. Doi:10.1089/Gte.2006.10.186
Carcavilla, A., Santome, J. L., Pinto, I., Sanchez-Pozo, J.,
Guillen-Navarro, E., Martin-Frias, M., . . . Ezquieta, B.
(2013). Leopard Syndrome: A Variant Of Noonan
Syndrome Strongly Associated With Hypertrophic
Cardiomyopathy. Rev Esp Cardiol (Engl Ed), 66(5),
350-356. Doi:10.1016/J.Rec.2012.09.015
Carta, C., Pantaleoni, F., Bocchinfuso, G., Stella, L., Vasta,
I., Sarkozy, A., . . . Tartaglia, M. (2006). Germline
Missense Mutations Affecting Kras Isoform B Are
JIMC 2020 - 1’s t Jenderal Soedirman International Medical Conference (JIMC) in conjunction with the Annual Scientific Meeting
(Temilnas) Consortium of Biomedical Science Indonesia (KIBI )
328

Associated With A Severe Noonan Syndrome
Phenotype. Am J Hum Genet, 79(1), 129-135.
Doi:10.1086/504394
Cessans, C., Ehlinger, V., Arnaud, C., Yart, A., Capri, Y.,
Barat, P., . . . Edouard, T. (2016). Growth Patterns Of
Patients With Noonan Syndrome: Correlation With
Age And Genotype. Eur J Endocrinol, 174(5), 641-650.
Doi:10.1530/Eje-15-0922
Chinton, J., Huckstadt, V., Moresco, A., Gravina, L. P., &
Obregon, M. G. (2019). Clinical And Molecular
Characterization Of Children With Noonan Syndrome
And Other Rasopathies In Argentina. Arch Argent
Pediatr, 117(5), 330-337.
Doi:10.5546/Aap.2019.Eng.330
Cirstea, I. C., Kutsche, K., Dvorsky, R., Gremer, L., Carta,
C., Horn, D., . . . Zenker, M. (2010). A Restricted
Spectrum Of Nras Mutations Causes Noonan
Syndrome. Nat Genet, 42(1), 27-29.
Doi:10.1038/Ng.497
Kobayashi, T., Aoki, Y., Niihori, T., Cave, H., Verloes, A.,
Okamoto, N., . . . Matsubara, Y. (2010). Molecular And
Clinical Analysis Of Raf1 In Noonan Syndrome And
Related Disorders: Dephosphorylation Of Serine 259
As The Essential Mechanism For Mutant Activation.
Hum Mutat, 31(3), 284-294. Doi:10.1002/Humu.21187
Lepri, F., De Luca, A., Stella, L., Rossi, C., Baldassarre, G.,
Pantaleoni, F., . . . Tartaglia, M. (2011). Sos1 Mutations
In Noonan Syndrome: Molecular Spectrum, Structural
Insights On Pathogenic Effects, And Genotype-
Phenotype Correlations. Hum Mutat, 32(7), 760-772.
Doi:10.1002/Humu.21492
Lepri, F. R., Scavelli, R., Digilio, M. C., Gnazzo, M.,
Grotta, S., Dentici, M. L., . . . Dallapiccola, B. (2014).
Diagnosis Of Noonan Syndrome And Related
Disorders Using Target Next Generation Sequencing.
Bmc Med Genet, 15, 14. Doi:10.1186/1471-2350-15-
14
Roberts, A. E., Araki, T., Swanson, K. D., Montgomery, K.
T., Schiripo, T. A., Joshi, V. A., . . . Kucherlapati, R. S.
(2007). Germline Gain-Of-Function Mutations In Sos1
Cause Noonan Syndrome. Nat Genet, 39(1), 70-74.
Doi:10.1038/Ng1926
Alteration of the Signal Transduction Pathway in RASopathies as a Basis of Targeted Therapeutic Drug Development
329
