
Efficient Analysis of Homeostasis of Gene Networks with Compositional
Approach
Sohei Ito
1,2
, Kenji Osari
3
, Shigeki Hagihara
4
and Naoki Yonezaki
5,6
1
Department of Fisheries Distribution and Management, National Fisheries University, 2-7-1 Nagata-Honmachi,
Shimonoseki, Yamaguchi, Japan
2
School of Business Administration in Karvin´a, Silesian University in Opava, Univerzitn´ı n´am. 76,
733 40 Karvin´a, Czech Republic
3
Yahoo Japan Corporation, 9-7-1 Akasaka, Minato-ku, Tokyo 107-6211, Japan
4
Department of Computer Science, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan
5
Graduate School of Information E nvironment, Tokyo Denki University, 2-1200, Muzai Gakuendai,
Inzai-shi, Chiba 270-1382, Japan
6
The Open University of Japan, 2-11 Wakaba, Mihama-ku, Chiba City,
Chiba 261-8586, Japan
Keywords:
Gene Regulatory Network, Systems Biology, Homeostasis, Temporal Logic, Realisability.
Abstract:
Homeostasis is an important property of life. Thanks to this property, living organisms keep their cellular
conditions within an acceptable range to function normally. To understand mechanisms of homeostasis and
analyse it, the systems biology approach is indispensable. For this purpose, we proposed a qualitative approach
to model gene regulatory networks with logical formulae and formulate the homeostasis in terms of a kind
of logical property – called realisability of l inear temporal logic. This concise formulation of homeostasis
naturally yields the method f or analysing homeostasis of gene networks using realisability checkers. However,
the realisability problem is wel l-known for its high computational complexity – double-exponential in the size
of a formula – and the applicability of this approach will be limited to small gene networks, since the size
of formula increases as the network does. To overcome this limitation, we leverage a compositional method
to check realisability in which a formula is divided into a few sub-formulae. The difficulty in compositional
approach is that we do not know how we obtain a good division. To tackle this issue, we introduce a new
clustering algorithm based on a characteristic function on formulae, which calculates the size of formulae and
the variation of propositions. The experimental results show that our method gives a good division to benefit
from the compositional method.
1 INTRODUCTION
Since the end of the 20th century, the advances of ex-
perimental and high-throughpu t technologies in mo-
lecular biology have enabled us to produce huge
amount of biological data. Compared to such advan-
cement, the understanding of biolo gical systems and
how th ey work seems less understood than expected.
As genes an d proteins are components of the system,
knowledge about each component does not give us
how the system is working as entirety. Therefore , a
new research field - systems biology - has emerged.
Systems biology aims to understan d organisms as
systems. Systems biology covers many topics such
as structural identification of biological systems, con-
struction of mathematical models which fit to obser-
ved phenom ena, and development of modelling and
analysing tools (Funahashi et al., 2003; Naldi et al.,
2009; Helikar et al., 2012).
Usual approach in systems biology is to model
a system with ordinary differential equations. Since
there is uncertainty in kinetic pa rameters, we fit them
to observed data to obtain a plausible model. Ho-
wever, the more comp onents we have in the system,
the more equations w e have thus parameter fitting
or computing analytical solution of the equations be-
come almost infeasible. To overcome this difficulty,
computational models have been used in systems bi-
ology (Fisher and Henzin ger, 2007), which abstract
real numer ic al behaviours into some discrete state se-
quences. There are several approaches to such com -
putational models depending on underlying forma-
Ito S., Osari K., Hagihara S. and Yonezaki N.
Efficient Analysis of Homeostasis of Gene Networks with Compositional Approach.
DOI: 10.5220/0006093600170028
In Proceedings of the 10th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2017), pages 17-28
ISBN: 978-989-758-214-1
Copyright
c
2017 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
17

lisms; Boolean networks (Thomas, 1991), Petri nets
(Heiner et al., 2008 ), timed-automata (Batt et al.,
2007) and process algebras (Ciocchetta and H illston,
2009). The limitation of such computational models
is that they re quire concrete mo le cular me chanisms,
regulatory logics and sometimes a kinetic parameters.
Since biological systems are incomple te in most ca-
ses, the applicability of such computational models
are limited. In this regard, the constra int-based mo-
delling is promising in which systems are described
as a set of constraints (Palsson, 2000). We can mo del
biological systems with incomp lete information. The
more information we have on the target system, the
more constraints we have in a model.
The constraint-based modelling paradigm has a
good connection with lo gical spec ification of rea c tive
systems in whic h the system behaviour is described as
a set of logical constraints (Mori and Yonezaki, 1993;
Vanitha et al., 2000; Osari et al., 2014). Based on
this close connection, we proposed a qualitative met-
hod for modelling and analysing gene networks (Ito
et al., 2010; Ito e t al., 2015b) using linear temporal
logic (LTL). In this approach, we specify possible be-
haviours of networks by LTL as a set of logical con-
straints an d analyse properties of networks (also w rit-
ten in LTL) by checking satisfiability of the formulae.
This idea is based on the verification of reactive sy-
stem specifications. Using this correspondence, we
later formulated the notion of homeostasis by reali-
sability of reactive system specifications (Pnueli and
Rosner, 1989; Abadi et al., 198 9) and demonstrated
how homeostasis in gene networks can be analysed
(Ito et al., 2014a).
The problem in our approach was the high-
complexity of checking LTL realisability which is
doubly-exponential in the size of a formula. As we
reported in (Ito et al., 2015a), c hecking hom e ostasis
of gene ne tworks with even a few nodes takes several
minutes. Since the sizes of formulae characterising
possible behaviours of a network is proportional to
the size of the network, we need to devise a me thod
to mitigate the computational difficulty in th e an alysis
of homeostasis.
To facilitate realisability checking, a compositio-
nal approach has been a promising method to analyse
large for mulae (Filiot et al., 2011) in which an LTL
formu la is d ivided into several sub-formulae that are
analysed separately. The results are the n merged to
obtain the final outcome of the verification. The non-
trivial p roblem in compositiona l analysis is how to di-
vide a formula, which is critical to the perf ormance of
analysis.
The aim of this paper is to provide a method for
finding good divisions as a front-end of a compo-
sitional analysis of homeostasis. Our approach to
this problem is to develop a new clustering algorithm
which clusters clauses (a piece of formula) based on
a ‘score’ of clusters. The ‘score’ of clusters indicates
how the clustering is good for compositional analysis.
We implement our algorithm and show experimental
results of analysing homeostasis of several gene net-
works. For larger gene networks, the gain from com-
positional approach is quite much: with compositi-
onal approach with our clustering front-en d, we can
verify in a few minutes, or even in a few seconds, net-
works that cannot be verified with monolithic appro-
ach in 1 hour.
The rest of the paper is o rganised a s follows.
Section 2 introduces LTL and the notion of realisa-
bility of reactive system specifications. Section 3 re-
views how we model gene networks in LTL and for-
mulates homeostasis by realisability. Section 4 intro-
duces the compositional analy sis of realisability. We
show how we divide a network specifications to com-
positionally ana lyse homeostasis. Section 5 shows
and discusses the experime ntal results of compositi-
onal analysis. In section 6, we discuss the relations-
hip between our m ethod and other similar approaches.
Section 7 offers conclusion and discusses some future
directions.
2 PRELIMINARY
2.1 Linear Temporal Logic
If A is a finite set, A
ω
denotes the set of all infinite
sequences on A. The i-th element of σ ∈ A
ω
is deno-
ted by σ[i]. Let AP be a set of propositions. A time
structure is a sequence σ ∈ (2
AP
)
ω
where 2
AP
is the
powerset of A P. The formulae of LTL are defined as
follows.
• p ∈ AP is a formula.
• If φ and ψ are for mulae, then ¬φ,φ∧ ψ,φ∨ ψ and
φUψ are also formulae.
We introduce the usual abbreviations: ⊥ ≡ p ∧ ¬p
for some p ∈ AP, ⊤ ≡ ¬⊥, φ → ψ ≡ ¬φ ∨ ψ, φ ↔
ψ ≡ (φ → ψ) ∧ (ψ → φ), Fφ ≡ ⊤Uφ, Gφ ≡ ¬F¬φ,
and φW ψ ≡ (φU ψ) ∨ Gφ. We assume that ∧, ∨ and
U binds more strongly than → and unary connectives
binds more strongly th an binary ones.
Let σ be a time structure and φ be a formula. The
semantics of LTL is defined by the relation σ |= φ re-
presenting φ is true in σ. The satisfaction relation |=
is defined as follows.
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
18

σ |= p iff p ∈ σ[0] for p ∈ AP
σ |= ¬φ iff σ 6|= φ
σ |= φ ∧ ψ iff σ |= φ and σ |= ψ
σ |= φ ∨ ψ iff σ |= φ or σ |= ψ
σ |= φUψ iff (∃i ≥ 0)(σ
i
|= ψ and
∀ j(0 ≤ j < i)σ
j
|= φ)
where σ
i
= σ[i]σ[i + 1].. . , i.e. the i-th su ffix of σ.
An LTL formula φ is satisfiable if there exists a time
structure σ such that σ |= φ. We a lso say that σ is a
model of φ.
2.2 Reactive Systems and Realisability
A reactive system is defined as a triple hX ,Y,ri, whe re
X is a set of events caused by the environment, Y is a
set of events caused by the system and r : (2
X
)
+
→ 2
Y
is a reaction function. Th e expression (2
X
)
+
denotes
the set of a ll finite sequences on subsets of X. A re-
action function determines how the system reacts to
environmental (or external) input sequences. Reactive
system is a natural formalisation o f systems which ap-
propriately respond to requests from the environment.
Systems controlling vending machines, elevators, air
traffic and nuclear power pla nts are examples of re-
active systems. Gene networks which respond to in-
puts or stimulation from the environme nt such as glu-
cose increase, change of temperature or blood pres-
sure can also be considered as reactive systems.
A specification of a reactive system stipulates how
it responds to inputs from the environment. For exam-
ple, for a controller of an elevator system, a specifica-
tion will be e.g. ‘if the open button is pushed, the door
opens’ or ‘if a call button of a certain floor is pushed,
the lift will come to the floor’.
Realisability is an important property of reactive
system specifications (Pnueli and Rosner, 1989;
Abadi et al., 1989). A realisable specification guaran-
tees that there exists a r eactive system wh ich respo nds
to any environmental inpu t of any timing without vi-
olating the specification.
To check realisability of a reactive system spe-
cification, it should be described in a language with
formal and rigorous semantics. LTL is known to be
one of many other formal lang uages suitable for this
purpose and several realisability checkers of LTL are
available (Jobstmann et al., 2007; Filiot et al., 2 009;
Bloem et al., 2010).
Now we define the notion of realisability of LTL
specifications. Let AP be a set of atomic propositions
which is partitioned into X and Y . X corr esponds to
external events and Y to internal events. We denote
a time structure σ on AP as hx
0
,y
0
ihx
1
,y
1
i. .. where
x
i
⊆ X, y
i
⊆ Y and σ[i] = x
i
∪ y
i
. Let φ be an LTL
specification. We say hX,Y,φi is realisable if there
x y
+
-
+
Figure 1: A gene network in which x and y are ge-
nes. Plus-edges represent activation relationship and
minus-edges represent inhibition relationship. Ge ne x
receives positive input from environment.
exists a reactive system RS = hX ,Y, ri such that
∀ ˜x.behave
RS
( ˜x) |= φ,
where ˜x ∈ (2
X
)
ω
and behave
RS
( ˜x) is the infinite beha-
viour determined by RS, that is,
behave
RS
( ˜x) = hx
0
,y
0
ihx
1
,y
1
i. .. ,
where ˜x = x
0
x
1
.. . and y
i
= r(x
0
.. .x
i
).
Intuitively a specification φ is realisable if there
exists a system which controls its internal events in
reaction to any sequence of external events, so that its
behaviour satisfies the specification φ.
3 QUALITATIVE ANALYSIS OF
HOMEOSTASIS IN GENE
NETWORKS
In this section we review how we model the gene net-
works and analyse homeostasis of them by LTL.
3.1 Modelling Possible Behaviours of
Gene Networks in LTL
Qualitative pr inciples for characterising behaviours of
a gene network are as follows:
• A gene is ON when its activators are expressed
beyond so me thresholds.
• A gene is OFF when its inhibitors are expressed
beyond so me thresholds.
• If a gene is ON, its expression level increases.
• If a gene is OFF, its expression level decreases.
To form a lly describe these principles in LTL, we in-
troduce propositions for a given network which repre-
sent the state (or c onfigu ration) of a network, such as
whether genes are ON and whether gene s are expres-
sed beyond c ertain thresholds.
We show how we describe behaviours of networks
using an example network depicted in Fig. 1, where
x and y repre sent gene s, p lus-edges mean activation
and the minus-edge inhibition. Since gene x activates
gene y, there exists a threshold expression level of x
Efficient Analysis of Homeostasis of Gene Networks with Compositional Approach
19

above which gene x activates gene y. We write x
y
for
this threshold level. Similarly, gene y has the thres-
hold y
x
to inhibit x. Moreover, gen e x receives the
positive input from environment. We wr ite e
x
for the
threshold of the input to activate x.
For th is network we introduce th e following pro-
positions.
• on
x
, on
y
: whether gene x and y are ON r especti-
vely.
• x
y
, y
x
: whether gene x and y are expressed beyond
the threshold x
y
and y
x
respectively
1
.
• in
x
: whether the input to x is ON.
• e
x
: whethe r the positive input from the environ-
ment to x is beyond the threshold e
x
.
Using these propositions, we specify the above
qualitative principle s in LTL. For example, the fact
‘gene y is positively regulated by gene x’ is descr ibed
as
G(x
y
↔ on
y
)
in LTL. Intuitively this formula says gene y is ON if,
and only if, gene x is expressed beyond the threshold
x
y
(i.e. proposition x
y
is true) due to positive effect of
gene x toward gene y. As for gene x, it is negatively
regulated by gene y and has positive inp ut from the en-
vironm ent. A co ndition for activation and inactivation
of such multi-regulated genes depends on a function
which merges the multiple effects. We assume that
gene x is ON if gene y is not expressed beyon d the
threshold y
x
and, in addition, the input from the envi-
ronment to gene x is beyo nd the threshold e
x
; that is,
the negative e ffect of gene y is not opera ting and the
positive effect of the input is ope rating. Then this can
be described as
G(e
x
∧ ¬y
x
→ on
x
).
This formula says that if the input level is beyond the
threshold e
x
and gene y is not exp ressed beyond the
threshold y
x
(i.e. proposition y
x
is false; ¬y
x
is true),
then gene x is ON.
If gene x is ON, the exp ression level of gene x is
growing. In other words, it will reach the threshold x
y
unless gene x becomes OFF prematurely. This fact is
described as
G(on
x
→ F(x
y
∨ ¬on
x
)).
If g e ne x is ON and expressed beyond the threshold
x
y
, it keeps the level until gene x is OFF since this is
the largest threshold of gene x. This can be described
as
G(on
x
∧ x
y
→ x
y
W ¬on
x
).
1
Note that the threshold levels are denoted in roman
whereas the propositions corresponding to them are denoted
in italic.
This formula means ‘if gene x is ON and the current
expression level of gene x is above x
y
, gene x keeps
its level as long as gene x is ON’.
If gene x is OFF, its transcription produ c t decrea-
ses due to degradation. We have the symmetric for-
mulae to the case of gene x being ON:
G(¬on
x
→ F(¬x
y
∨ on
x
)),
G(¬on
x
∧ ¬x
y
→ ¬x
y
W on
x
).
We have similar c la uses for expression of gene y.
We do not th oroughly explain how w e d escribe
constraints which model the possible behaviours of
gene network s. Interested readers may wish to con-
sult (Ito et al., 2 015b) for detail.
3.2 Analysing Homeostasis by
Realisability Checking
Biological homeostasis is informally defined as the
tendency of a system to maintain its internal stabi-
lity or f unction against any situation or stimulus. This
property is closely related to realisability since it says
that there is a system which responds to any environ-
mental inputs of any timing while keeping its speci-
fied internal conditions. Using this correspondence
we for mulate homeostasis by realisability.
In the previous section, we showed that a gene net-
work can be modelled by an LTL formula which is the
conjunc tion of clau ses. Precisely speaking, the speci-
fication of a given network is a triple hE, I, ϕi where
• E: the set of external propositions (inputs),
• I: the set of internal propositions (genes and thres-
holds),
• ϕ: the formula which characterises the possible
behaviours of a given network.
We also write a biological property or function of
the network, which we are to check whether the given
network maintains it in response to any external input
sequence. The examples of such property will be ‘the
expression level of a certain gene is within a to le rable
range’, ‘once a gene becomes ON, its expression will
be suppressed afterwards’, et cetera. Such properties
can be easily described in LTL.
In realistic situation, we sometimes put an as-
sumption (or constraints) on the environment which
restricts the in put sequ ence. For example, we may
consider the case that an input is oscillating, an input
is always coming, or a certa in input com es only after
another comes, and so on. Sometime s it is useful to
consider homeostasis of a system under such environ-
mental assumptions. We also describe suc h assumpti-
ons in LTL.
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
20

Now we define homeostasis of a gene network
with respect to a p roperty under an environmental as-
sumption. The following definition is an extended
version of our previous definition of homeostasis (Ito
et al., 201 4a) in that we include environmental as-
sumption ξ.
Definition 1. A property ψ is homeostatic with re-
spect to a behaviour specification hE, I, ϕi u nder the
environmental assumption ξ if hE, I, ξ → ϕ∧ ψi is re-
alisable.
Note that a homeostasis of a gene network is de-
fined with a certain bio logical property. A network
which is homeostatic with r espect to a certain pro-
perty has a strategy to respond for any input sequence
which satisfies ξ, witho ut violatin g neither behaviour
constraint ϕ nor the property ψ. We can set ξ as true
(i.e. empty clause), which amounts to put no assump-
tion on environment.
Example 1. The network depicted in Fig. 1 is ho-
meostatic with respect to a p roperty ‘whenever gene x
becomes ON, the expression of gene x will be suppres-
sed afterwards’. This property is described in LTL as
follows:
G(on
x
→ F(¬on
x
∧ ¬x
y
)).
Let ϕ be a behaviou ral specification of the network
(partly shown in the previous section). This is ve-
rified by checking realisability of the specification
h{in
x
}, {on
x
,on
y
,x
y
,y
x
,e
x
}, ϕ ∧ G(on
x
→ F(¬on
x
∧
¬x
y
))i.
This property is also homeostatic if we put an en-
vironm e ntal assumption ‘the input to x is always ON’
which is described as:
Gin
x
.
That is to say, the specification
h{in
x
}, {on
x
,on
y
,x
y
,y
x
,e
x
}, Gin
x
→ ϕ ∧ ψi is re -
alisable (where ψ represents the above property).
Since we de fined homeostasis by realisability, we
can use realisability checkers to conduct such ana-
lysis. A n obstacle with this framework is the h igh-
complexity of LTL realisability problem which is
2EXPTIME- c omplete in the size o f a formula (Pnu-
eli and Rosner, 1989). Since the size of a network
specification is proportional to the size of a network,
the analysis of a larger network will be intractable in
general. The next section discusses the compositional
analysis method to mitigate this difficulty.
4 COMPOSITIONAL ANALYSIS
OF HOMEOSTASIS
Recent advances in realisability checking technique
enable us to handle large specifications. Since our
framework to analyse homeostasis uses realisability
checking, we can re ap the benefit from the advan-
ces. Here we leverage the compositional algorithm
in realisability checking (Filiot et al., 2011). In our
setting, the com positional analysis of homeostasis of
gene networks is stated as fo llows:
1. For a given specification hE, I, ξ →
V
1≤i≤n
ϕ
i
i,
compute a ‘good’ clustering of the formula
{c
1
,. .. , c
k
}, where each c
ℓ
consists of ϕ
i
s, e.g.
network specifications and bio logical properties.
2. We check the realisability of each sub-
specification
hE, I, ξ →
V
ϕ∈c
ℓ
ϕi(ℓ ∈ {1,. .. ,k}).
3. We merge the r esult of individual results.
Step 2 and 3 can be automatically done by the tool
Acacia+ (Boh y et al., 2 012). The problem is how
we obtain a ‘good ’ clustering of a given specification.
Before proceedin g, we first review the algorithm of
Acacia+ to solve realisability compositionally.
4.1 Compositional Approach to
Realisability Problems
The algorithm implemented in Acacia+ to solve rea-
lisability of LTL formula ϕ is as follows:
1. Translate ϕ to a universal co-B¨uchi a utomaton A
ϕ
.
2. Translate A
ϕ
to a safety game G(A
ϕ
).
3. Compute the winning strategy of the game G(A
ϕ
).
It is reported that th e first step, i.e. translating an
LTL formula to a n equivalent automaton, is the bott-
leneck of the algorithm. Known algorithms to trans-
late LTL formulae into equivalent automata run in ex-
ponen tial time with respect to the sizes of formulae.
Especially for large LTL formulae the first step do es
not finish. To overcome this p roblem, the compositi-
onal approach is proposed. Large LTL formulae are
often written as ϕ = ϕ
1
∧ ϕ
2
∧ ··· ∧ ϕ
n
. Thus in com-
positional appro ach, each sub-formula ϕ
i
is transla-
ted into autom aton A
ϕ
i
and thus translated into a local
game G(A
ϕ
i
), then the winning strategy is computed
for each G(A
ϕ
i
). Thanks to the nice property of sa-
fety game s, the winning strategy fo r G(A
ϕ
), the stra-
tegy for the original g ame, can be computed from the
winning strategies for each local game G(A
ϕ
i
).
In general, the specification is of the form ξ → ϕ
where ξ is an environmental assumption. Therefore ξ
Efficient Analysis of Homeostasis of Gene Networks with Compositional Approach
21

is distributed to each ϕ
i
, that is, we solve the realisa-
bility of each ξ → ϕ
i
separately.
The m e rit of this compositiona l approach is that
we c an reduce the time of translating formulae into
equivalent auto mata, since eac h ϕ
i
is much smal-
ler than the original formula ϕ. Note that, however,
we have extra cost of merging the winning strategies
for local games, compared to non-compositional (i.e.
monolith ic ) approach. For compositional approach to
be u seful, the reduction in automata translation must
prevail the additional cost of merging the local win-
ning strategies. Thus the performance of compositio-
nal approac h depend s on how we divide ϕ into several
ϕ
i
s.
4.2 Applying to Our Problem
Since behaviour sp e cifications for g ene ne tworks are
written as conjunctions of clauses (see section 3.1),
we can apply the compositional app roach introduced
in the previous section. Our specification in gene-
ral consists of many clauses, thus it is not efficient
to solve every single clauses sepa rately as the over-
head of integrating the winning strategies of the local
games increases according to the num ber of local ga-
mes.
Thus the possible approach is to distribute the
clauses in to several clusters c
1
,c
2
,. .. , c
k
, i.e. ϕ =
(
V
c
1
) ∧ (
V
c
2
) ∧ ·· · ∧ (
V
c
k
) where
V
c
i
=
V
φ∈c
i
φ.
The problem is how to distribute them. What is a good
clustering?
For example, suppose that we have 20 clauses and
distributing them into 2 clusters. It seems better to put
10 clauses into ea ch cluster than to put 1 clau se into
one cluster and 19 clauses into the other. In such une-
ven clustering, the 19 clauses cluster will be a bottle-
neck and the efficiency may not improve as a whole.
Thus a goo d clustering distributes clauses into clus-
ters as equally as possible. However, the number of
clauses may not be a good criterion, since the sizes
of form ulae are not the same; in an extreme situation,
the size of a ce rtain formula might be equal to the size
of the rest. Thus we distribute clauses into clusters
whose sizes are as equal as possible.
To facilitate winning strategy computation of each
local game, it is desirable to obtain small automata.
The size of automata A
ϕ
is determined by the number
of sub-formulae in ϕ. To compute the number of sub-
formu lae for each clause is not realistic because it is
exponential to the size of the formula. The plausible
criterion is the number of variation of propositions –
the more variation of propositions we have, the more
the number of sub-formulae. Thus we also take the
variation of propositions in a cluster into co nsidera-
tion, in addition to the size of a cluster.
Now we formalise the above idea. Let
C = {c
1
,. .. , c
k
} be a clustering o f a specification
hE, I, ξ →
V
1≤i≤n
ϕ
i
i, that is, each cluster c
ℓ
consists
of ϕ
i
s. Note that the environmental constraint ξ is co-
pied to each cluster. Let N(χ) and V (χ) respectively
denote the number and th e variations of propositions
in a formula χ. We introduce the evaluation function
F of clustering C as follows:
F (C) =
∑
1≤i≤k
E(ξ →
^
ϕ∈c
i
ϕ),
where E (χ) = N(χ)
2
+V (χ)
2
The fun ction E rep resents the evaluation of a single
formu la. Due to the squared terms in the function
E, we can easily see that a clustering consists of two
clusters of the sizes 5 and 5 is better than that of 9 an d
1. A good clusterin g is the one which minimises the
value of this function.
The number of possible clustering of n formulae
into k clusters is a S(n, k), i.e. the Stirling number
of the second kind. Thus the na¨ıve algorithm to find
the optimal cluster is not realistic in general. Thus we
introdu ce a suboptimal algorithm whose complexity
is O(kn
2
) which we show in Algorithm 1.
Algorithm 1: Suboptimal clustering algorithm.
Input: {ϕ
1
,. .. , ϕ
n
} (specification), k (the number of
the clusters)
Output: C = {c
1
,. .. , c
k
}
c
1
← {ϕ
1
,. .. , ϕ
n
}
for i = 2 to k do
c
i
←
/
0
end for
C ← {c
1
,. .. , c
k
}
min ← F (C)
repeat
for all ϕ ∈ c
1
do
change ← false
for i = 2 to k do
C
′
← {c
1
\{ϕ}, .. ., c
i
∪ {ϕ}, .. ., c
k
}
f ← F (C
′
)
if f < min then
C ← C
′
min ← f
change ← true
end if
end for
end for
until change = false
This algorithm starts with the cluster c
1
with
{ϕ
1
,. .. , ϕ
n
} and the others with empty sets. For each
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
22
step we move one ϕ in c
1
to another clu ster c
i
and
evaluate th e new clustering by function F . If the
evaluation decreases for some i, we adopt the mini-
mal one among such clusterings. We repeat this pr o-
cess until moving ϕ from c
1
to any other clusters no
longer decreases the evaluation. Since the outermost
loop is executed at most n times, and each step de-
crements the number of elements of c
1
, this a lgorithm
runs in time O(kn
2
). We implemented the algorithm
in OCaml and confirme d that this suboptimal algo-
rithm computes a clu stering whose evaluation is as
good as the optimal clustering.
5 EXPERIMENTAL RESULTS
AND DISCUSSION
This section discusses the expe rimental results of ho-
meostasis analysis pe rformed on several networks.
Since comp ositional analysis is available for only re-
alisable specifications, all specifications used in this
experiment are realisable.
To solve the realisability of an LTL spe cification
ϕ, we fir st construc t an ω-automa ton which is equi-
valent to ϕ, then construct a game on the automaton
and compute a winning strategy of the system which
can respon d any environm ental req uests. Co mposi-
tional analysis decreases the cost o f constructin g ω-
automata from LTL formulae since the form ula is di-
vided into several small sub-formulae. However, we
have extra cost of merging all winning strategies com-
puted for each sub-formulae. Therefore , increasing
the number of clusters ma y not ne c essarily reduce the
cost of analysis as a whole. However, to predict the
optimal number of clustering a priori is difficult. We,
hence, demonstrate our clustering meth od with diffe-
rent number of clusters to see the impact of the num-
ber of clusters in our compositional method.
x y
+
+
-
Figure 2: A bistab le switch ( Ito et al., 2014a).
x y
+
+
- -
Figure 3: A bistable switch with a dditional inputs (Ito
et al., 2014a).
The result of experiments are shown in table 1.
These experiments are car ried out on a computer with
Intel Core i7-3820 3.60GHz CPU and 32GB memory.
The realisability checker used is Acacia + (Bohy et a l.,
2012). The network ‘bistable switch’ is the network
that consists of two genes x and y where x activates y,
y activates x and x receives negative input from envi-
ronment (Fig. 2). The network ‘bistable switch2’ is
the same as ‘bistable switch’ except both gene x and
y receive negative inputs from environment (Fig. 3).
The networks ‘anti-stress response (a )(b)(c)’ are th e
stress response networks studied in (Zhang and An-
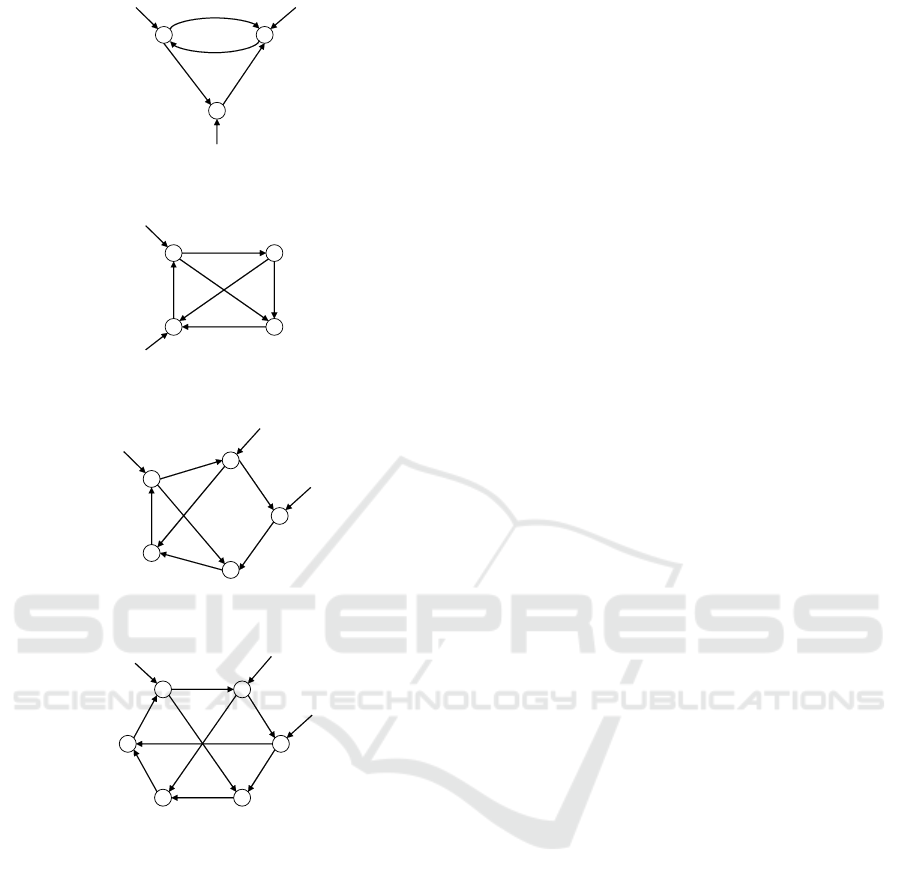
dersen, 2007) (Fig. 4). The networks ‘3 genes’ to ‘6
genes’ are de picted in Fig s. 5 to 8. The homeosta-
tic properties we analysed are tendency to ease stress
(described as G(stress → F¬stress)) for anti-stress re-
sponse networks and stability of a certain gene ex-
pression (described as Gon) for others. A s environ-
mental assumptions, we put every input is oscillating;
it is written as G((inp ut → F¬input) ∧ (¬input →
Finput)).
As we can see from the result, we benefit from
compositional analyses compared to the monolithic
approa c h (k = 1). Th e improvements in analyses of
specifications from ‘3 genes’ to ‘6 genes’ are remar-
kable.
Concernin g the number of clusters, as we stated
at the beginning of this section, incre asing the num -
ber of clusters does not necessarily reduce the entire
verification time. We can see it from ‘anti-stress re-
sponse(c)’ wher e the cases for k > 4 are worse than
the monolithic approach. Even for larger specificati-
ons, the best number of clusters seems to be 2 or 3.
The computational time of our clustering algo-
rithm is less than 0.01 regardless of the number of
clusters for a ll networks excep t for ‘6genes’ at k =
6 (0.11 seconds). This means that we can ignore
the cost of computing clusters to use compositional
analysis even for small networks such as ‘bistable
switch’. Note that table 1 includes the clustering ti-
mes.
Now we compare our m ethod to ran dom cluste-
ring. For each specification, we generate 50 random
clusterings. We first randomly choose the size (i.e. the
number of clauses) of each cluster, then we randomly
distribute clauses to each cluster accordin g to the size.
We show the results o f random clustering in table
2. When calculating average time of verification, the
clusterings which cannot be verified within 1 hour are
treated as 1 hour. The mark ⋆ in the table shows that
there were suc h clusterings. Thus the true average is
greater than the shown value.
For larger spec ifica tions such as ‘bistable
switch2’, ‘3genes’, ‘4genes’ and ‘5genes’, we can
clearly see that our method outper forms random clus-
tering. Readers might notice that for the network of
Efficient Analysis of Homeostasis of Gene Networks with Compositional Approach
23

-+
--
+
++
+-
-
(a)
(b) (c)
-+
+
+
Figure 4: Anti-stress networks.
Table 1: Experimental results. Columns ‘E’ and ‘I’ respectively show the numb e rs of external propositions and
internal propositions. Column ‘S’ shows th e size of formula. (i.e. numbe r of connectives and propositions).
The columns ‘Time(s)’ show the total time of the verification for various number of clusters (k). ‘k = 1’ means
non-compositional ana lysis. For ‘k > 2 the times include computation of cluster ing.
Time(s)
Network E I S k = 1 k = 2 k = 3 k = 4 k = 5 k = 6
bistable switch 1 6 232 0.40 0.07 0.08 0.10 0.10 0.07
bistable switch2 2 8 353 206.81 0.81 0.41 0.42 0.51 0.53
anti-stress response (a) 1 9 289 0.48 0.31 0.35 0.41 0.47 0.51
anti-stress response (b) 1 7 237 0.22 0.12 0.13 0.15 0.16 0.18
anti-stress response (c) 1 9 288 0.39 0.31 0.38 0.41 0.47 0.51
3 genes 3 10 418 1617.87 5.03 5.43 6.08 5.82 6.30
4 genes 2 12 444 > 3600 4.07 4.48 5. 16 5.69 6.21
5 genes 3 15 547 > 3600 173.31 123.80 192.17 135.77 194.75
6 genes 3 18 646 > 3600 2676.37 2792.61 2877.03 2942.62 3087.91
Table 2: Results of random clustering. It shows average verification times of 50 randomly clustered specs. The
figures show the time (in seconds) to check homeostasis. The stars on the figures show that some trials failed due
to time out of 1 hour. Such clusterings are tr eated as finished in 3600 seconds for the sake of expedience.
Network k = 2 k = 3 k = 4 k = 5 k = 6
bistable switch 0.11 0.11 0.11 0.11 0.12
bistable switch2 12.20 4.39 2.24 1.62 1.23
anti-stress response (a) 0.36 0.38 0.43 0.44 0.47
anti-stress response (b) 0.13 0.14 0.15 0.16 0.17
anti-stress response (c) 0.35 0.37 0.40 0.43 0.46
3 genes 81.80 29.44 50.88 10.44 10.94
4 genes 687.47
⋆
441.53
⋆
166.43 45.99 130.66
5 genes 1746.24
⋆
845.6
⋆
772.39
⋆
841.6
⋆
842.74
⋆
6 genes 3220.6
⋆
2953.01
⋆
2767.81
⋆
2830.68
⋆
2641.21
⋆
‘6gene s’, the random clustering seems to ou tperform
our method for some k. However, this is because
we treated timed-out samples as 3600 seconds when
averaging the results. Though we do not k now the
true average, it will be worse than the resu lts of ou r
method.
For smaller specifications, such a s ‘bistable
switch’ and ‘anti-stress response’ networks, our met-
hod is no t clearly better than r andom clustering. The
reason is that the formulae are not so large that the
automata construction is not a heavy task in the veri-
fication process. For such specifications our strategy
– to decrease the time of automata construction – may
not be suitable.
Readers might wonder why ‘4genes’ for k = 5 is
considerably smaller than others in r a ndom cluste-
ring. Plausible expla nation is that the random sam-
pling happened to give biased clusterings. What is
important here is that even for such biased random
sampling, our method is much better.
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
24
+
+
-
-
-
++
Figure 5: A ne twork consisting of 3 genes.
+
+
-
-
+
+
-
+
Figure 6: A ne twork consisting of 4 genes.
+
+
+
+
+
-
+
+
-
+
Figure 7: A ne twork consisting of 5 genes.
+
+
+
+
+
-
-
+
-
+
+
+
Figure 8: A ne twork consisting of 6 genes.
We also show the standard deviations of the
random clustering in table 3. As we can see from the
table, the standard deviations are hig h for larger spe-
cifications. This means that our evaluation function is
actually a statistically meaningful cr iterion, because
random clustering ranges from better to worse. Our
clustering algorithm tactfully chooses better ones.
We show another proof of the benefit of our al-
gorithm: relative standin gs of our results (the values
shown in Table 1) within the run-times of r andom
clusterings. In othe r words, the percentile r anks of
our results in the random clusterings. Tab le 4 shows
them. As we can see, for k = 2 or 3 our algorithm
gives good scores.
However, this table also shows that for k ≥ 4 our
evaluation function might not b e a good criterion.
This is because if we increase the number of clus-
ters, the size of for mulae in each cluster tends to be
small, so that the automata construction time decr e-
ases and becomes less im portant in the verification
process. Thus the larger k tends to impair the bene-
fit of our method. Rather, we on ly rea p the overhead
of me rging the winning strategies of the local games.
This suggests another tactics for clustering: we focus
on decreasing the computation time of winn ing stra-
tegy. We leave this issue for future work.
Before closing this section, we make a brief re-
mark on a treatmen t of unrealisable specifications. As
we men tioned at the beginning of this section, com-
positional appro ach is o nly applicab le for realisable
specification (Filiot et al., 2011; Bohy et al., 2012). In
the current implementation of Acacia+, a user choo-
ses options whether he checks realisability or u nreali-
sability (or both in parallel). Thus when we are to ve-
rify a specification for which we do not know whether
it is realisable or not, first we try realisability checking
compositionally. If the verifier cannot prove realisa-
bility, we try unrealisability checking monolithically.
However, since Acacia+ uses heuristic in which a cer-
tain bound is set when searching winning strategies,
sometimes neither realisability nor unrealisability is
proved. If this is the case, we set a larger bound and
repeat the process un til we have a definite result.
We summar ise this section. For larger sp ecificati-
ons, our method works well because the bottleneck in
the verification process is the automata construction.
If the n umber of clusters is small (2 or 3), ou r method
is especially effective. If we increase the number of
clusters, the bottleneck seems to shift from automata
construction to winning strategy compu ta tion. To in-
vestigate a method to reduc e the time for winning
strategy co mputation is an interesting future work.
6 RELATED WORK
Sensitivity or robustness of biological systems is close
but different from homeostasis. It analyses whe ther a
property holds for a similar degree/probability when a
parameter is mod ified, w hile homeostasis me ans that
a given property holds for a certain set of input se-
quences. (Rizk et al., 2009) de fines r obustness of a
biological system by measuring the deviation of the
‘satisfaction degree’ of an LTL formula (Fages and
Rizk, 2008) c a used by perturbations on parameters.
They can treat the initial value of an environmental
input as a parame ter, thus homeostasis is partially co-
vered. (Donz´e et al., 2011) is a similar approach to
(Rizk et al., 2009) for robustness analysis. They ana-
lyse continuous trajectories by means of signal tem-
Efficient Analysis of Homeostasis of Gene Networks with Compositional Approach
25
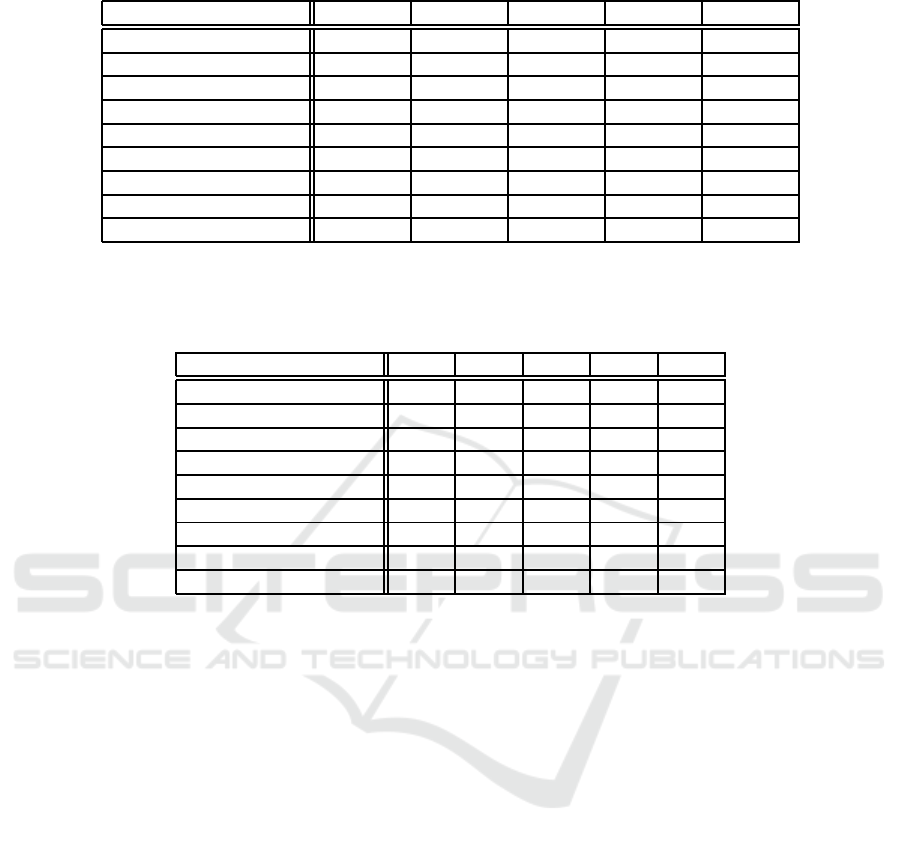
Table 3: Standard d eviation of the results of random clustering .
Network k = 2 k = 3 k = 4 k = 5 k = 6
bistable switch 0.05 0.03 0.02 0.01 0.01
bistable switch2 23.00 12.1 4.51 3.74 2.30
anti-stress response (a) 0.09 0.08 0.13 0.04 0.04
anti-stress response (b) 0.02 0.02 0.01 0.01 0.01
anti-stress response (c) 0.06 0.09 0.03 0.04 0.05
3 genes 183.01 138.24 273.49 13.04 22 .63
4 genes 1239.13
⋆
1021.42
⋆
618.16 218.94 377.97
5 genes 1593.59
⋆
1318.82
⋆
1183.29
⋆
1295.33
⋆
1270.92
⋆
6 genes 700.55
⋆
763.89
⋆
835.77
⋆
819.86
⋆
989.15
⋆
Table 4: Relative standing of the verification time with our method within the run-times of random clusterings.
The value ‘best’ means that our method was the best.
Network k = 2 k = 3 k = 4 k = 5 k = 6
bistable switch 10% 14% 39% 43% best
bistable switch2 38% 13% 15% 41% 41%
anti-stress response (a) 27% 17% 64% 88% 88%
anti-stress response (b) 31% 45% 70% 7 0% 92%
anti-stress response (c) 23% 86% 6 8% 92% 92%
3 genes 14% 26% 4 7% 27% 27%
4 genes 17% 8% 34% 38% 43%
5 genes 19% 12% 4 3% 15% 35%
6 genes 14% 26% 4 1% 44% 50%
poral logic (STL) and its satisfaction-degree. In their
approa c hes, it is unclear how to model arbitrary inpu t
scenario and assess the effect of it. SReach (Wang
et al., 2015) is a too l to analyse stochastic hybrid sys-
tems, which can be used to m odel biological systems.
SReach focuses the probabilistic reachability, that is,
a property whether a given system reaches safe (or
unsafe) states within a given probability range. SRe-
ach can conduct sensitivity analysis: Are the re sults
of reachability analysis the same for different p ossible
values of a certain system parameter? Since SReach
only considers probabilities on state transitions, it is
not clear how to model perturbations on inputs.
(Zhang and Andersen, 2007) analyses homeosta-
sis of anti-stress gene regulatory networks by means
of control theory. They consider stead y-state response
curves of anti-stress genes to a dose of stress. Their
control- theoretical method is tailored to anti-stress
networks and feasible to predict the effect of chan-
ging local r e sponse coefficients, but does not provide
a gene ric fr amework applicable to any network.
In contrast to above approaches, our approach is
purely qualitative. We do not ne e d any kinetic pa-
rameters, which is usually difficult to obtain. There
have been proposed several qualitative approa ches to
model and analyse biological systems based on se-
veral different fo rmalisms - BIOCHAM (Fages et al. ,
2004) based on rewriting system, SMBioNet (Ber-
not et al., 2004 ) and Qualitative networks (Schau b
et al., 2007) based on extended Boolean networks,
and GNA (de Jong et al., 2003) based on piecewise
linear differential equation. Compared to these for-
malisms, our LTL-based model of gene network s are
flexible and easy to construct since the basic principle
to model the behaviours are quite simple. Th e change
of condition or assumption (e.g. bias of multivariate
regulation) is im mediate. Although the above tools
are useful for checking wheth er a biological property
can be true in network behaviours, it is unknown how
to define and analyse homeostasis. As to the size of
analysable networks, we cannot directly compare th e
scalability since the properties ana lysed and behavi-
ours modelled in these tools are different and the cost
of analysis also depends on the complexity of the net-
work structure as well as the size. Fur thermore, the
examples demonstrated are very few and we do not
have canonical benchmarks in this field. For informa-
tion, we refer the size of networks analysed in these
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
26

tools; SMBioNet analy ses a 3 n odes network, GNA
a 10 nodes network and Qualitative networks a 2160
nodes network (in 10 hours). Qualitative network can
handle very large networks compared to others inclu-
ding our framework, but we should keep in mind that
Qualitative networks only consider pro positional pro-
perties on steady states of the network behaviour, i.e .
a temporal property such as ‘when a gene is activated
it will be suppressed later’ cannot be checked. Furt-
hermore, the behaviour of a Qualitative n etwork is de-
terministic. In contrast we consider all possible net-
work behaviours and their temporal properties with
responses to environmental inputs.
As for compositional analysis of gene networks,
we proposed to divide a network into several sub-
networks and verify them individually (Ito et al.,
2013; Ito et al., 2015b). T his means we manually di-
vide an LTL formula in to several sub-formulae, con-
sidering the network structure. We have not discussed
any algorithm to divide a network and the correspon -
ding fo rmula. In this paper, our method is based on
a syntactic structure o f a formula, not on a network
structure. Thu s it is easy to cluster a formula algo-
rithmically.
7 CONCLUSION
In this paper we proposed compositional analysis of
homeostasis of gene networks. We introduced a new
clustering algorithm based on char a cteristic function
on LTL formulae to divide a network sp ecification.
The experimental results show that our method dras-
tically imp roves the performance in analysing larger
network spec ifications. Our clustering algorithm is
not limited to analyse homeostasis of gene n etworks
but for r ealisability checking of any r eactive system
specification.
All examples in our experiments are car ried out
to see the impact of the sizes of networks. Th us the
homeostatic properties are simple and described as re-
latively small formulae compared to network specifi-
cations. We would like to assess the impact of homeo-
static properties to be more confident in our approach.
For furthe r improvement, we are interested in im-
porting Ito et al.’s approximate method (Ito et al.,
2014b) to check homeostasis of gene networks. Anot-
her important future direction is to analy se real net-
works w hich may now be feasible thanks to this work,
and elucidate regulation mechanisms contributing bi-
ological homeostasis.
REFERENCES
Abadi, M., Lamport, L., and Wolper, P. (1989). Reali-
zable and unrealizable specifications of reactive sys-
tems. In ICALP ’89: Proceedings of the 16th Interna-
tional Colloquium on Automata, Languages and Pro-
gramming, volume 372 of LNCS, pages 1–17, Lon-
don, UK. Springer-Verlag.
Batt, G., Salah, R. B. , and Maler, O. (2007). On timed
models of gene networks. In FORMATS 2007, volume
4763 of LNCS, pages 38–52.
Bernot, G., Comet, J., Richard, A., and Guespin, J. (2004).
Application of formal methods to biological regula-
tory networks: extending Thomas’ asynchronous lo-
gical approach with temporal logic. J. T heor. Biol.,
229(3):339–347.
Bloem, R., Cimatti, A., Greimel, K., Hofferek, G.,
K¨onighofer, R., Roveri, M., Schuppan, V., and See-
ber, R. (2010). RATSY – a new requirements analysis
tool with synthesis. In Proceedings of the 22nd inter-
national conference on Computer Aided Verification,
volume 6174 of LNCS, pages 425–429, Berlin, Hei-
delberg. Springer-Verlag.
Bohy, A., Br uy`ere, V., Filiot, E., Jin, N., and Raskin, J.-F.
(2012). Acacia+, a tool for LTL synthesis. In Procee-
dings of the 24th International Conference on Compu-
ter Aided Verification, CAV’12, pages 652–657.
Ciocchetta, F. and Hillston, J. (2009). Bio-PEPA: A fra-
mework for the modelling and analysis of biological
systems. Theor. Comput. Sci. , 410:3065–3084.
de Jong, H. , Geiselmann, J., Hernandez, G., and Page, M.
(2003). Genetic network analyzer: Qualitative simu-
lation of genetic regulatory networks. Bi oinformatics,
19(3):336–344.
Donz´e, A., Fanchon, E., Gatt epaille, L. M., Maler, O., and
Tracqui, P. (2011). Robustness analysis and behavior
discrimination in enzymatic reaction networks. PLOS
One, 6(9):e24246.
Fages, F. and Rizk, A. (2008). On temporal logic con-
straint solving for analyzing numerical data time se-
ries. Theor. Comput. Sci., 408(1):55–65.
Fages, F., Soliman, S., and Chabrier-Rivier, N. (2004). Mo-
delling and querying interaction networks in the bio-
chemical abstract machine BIOCHAM. J. Biol. Phys.
Chem., 4:64–73.
Filiot, E., Jin, N., and Raskin, J.-F. (2009). An antichain
algorithm for LTL realizability. In Proceedings of
the 21st International Conference on Computer Aided
Verification, volume 5126 of LNCS, pages 263–277,
Berlin, Heidelberg. Springer-Verlag.
Filiot, E., Jin, N., and Raskin, J.-F. (2011). Antichains and
compositional algorithms for LTL synthesis. Formal
Methods in System Design, 39(3):261–296.
Fisher, J. and Henzinger, T. (2007). Executable cell biology.
Nat. Biotechnol., 25(11):1239–1249.
Funahashi, A., Tanimura, N., Morohashi, M., and Kitano,
H. (2003). Celldesigner: a process diagram editor
for gene-regulatory and biochemical networks. BIO-
SILICO, 1:159–162.
Efficient Analysis of Homeostasis of Gene Networks with Compositional Approach
27

Heiner, M., Gilbert, D. R., and Donaldson, R . (2008). Petri
nets f or systems and synthetic biology. In SFM 2008,
volume 5016 of LNCS, pages 215–264.
Helikar, T., Kowal, B., McClenathan, S., Bruckner, M., R o-
wley, T., Madrahimov, A., Wicks, B., Shrestha, M.,
Limbu, K., and Rogers, J. A. (2012). The cell col-
lective: Toward an open and collaborative approach to
systems biology. BMC Systems Biology, 6(1):96.
Ito, S., Hagihara, S., and Yonezaki, N. ( 2014a). A qualita-
tive framework for analysing homeostasis in gene net-
works. In Proceedings of BIOINFORMATICS 2014,
pages 5–16.
Ito, S., Hagihara, S., and Yonezaki, N. (2015a). For-
mulation of homeostasis by realisability on linear
temporal logic. In Plantier, G., Schultz, T., Fred,
A., and Gamboa, H., editors, Biomedical Engineer-
ing Systems and Technologies: 7th International
Joint Conference, BIOSTEC 2014, Angers, France,
March 3-6, 2014, Revised Selected Papers, pages
149–164. Springer International Publishing, Cham.
http://dx.doi.org/10.1007/978-3-319-26129-4
10.
Ito, S., Ichinose, T., Shimakawa, M., Izumi, N., Ha-
gihara, S., and Yonezaki, N. (2013). Modu-
lar analysis of gene networks by linear tempo-
ral logic. J. Integrative Bioinformatics, 10(2).
http://dx.doi.org/10.2390/biecoll-jib-2013-216.
Ito, S., Ichinose, T., Shimakawa, M., Izumi, N., Hagi-
hara, S., and Yonezaki, N. (2014b). Formal ana-
lysis of gene networks using network motifs. In
Fern´andez-Chimeno, M., Fernandes, L. P., Alvarez,
S., Stacey, D., Sol´e-Casals, J., Fred, A., and Gam-
boa, H., editors, Biomedical Engineering Systems
and Technologies: 6th International Joint Confe-
rence, BIOSTEC 2013, Barcelona, Spain, February
11-14, 2013, Revised Selected Papers, pages 131–
146. Springer Berlin H ei delberg, Berlin, Heidelberg.
http://dx.doi.org/10.1007/978-3-662-44485-6
10.
Ito, S., Ichinose, T., Shimakawa, M., Izumi, N., Ha-
gihara, S., and Yonezaki, N. (2015b). Qualita-
tive analysis of gene regulatory networks by tem-
poral logic. Theor. Comput. Sci., 594(23):151–179.
http://dx.doi.org/10.1016/j.tcs.2015.06.017.
Ito, S., Izumi, N., Hagihara, S., and Yonezaki, N.
(2010). Qualitative analysis of gene regulatory net-
works by satisfiability checking of linear temporal lo-
gic. In Proceedings of BI BE 2010, pages 232–237.
http://dx.doi.org/10.1109/BIBE.2010.45.
Jobstmann, B., Galler, S., Weiglhofer, M., and Bloem, R.
(2007). Anzu: a tool for property synthesis. In Pro-
ceedings of the 19th international conference on Com-
puter aided verification, volume 4590 of LNCS, pages
258–262, Berlin, Heidelberg. Springer-Verlag.
Mori, R. and Yonezaki, N. (1993). Several realizability con-
cepts in reactive objects. In Information Modeling and
Knowledge Bases IV, pages 407–424.
Naldi, A., Berenguier, D., Faur´e, A., Lopez, F., Thieffry, D.,
and Chaouiya, C. (2009). Logical modelling of regu-
latory networks with ginsim 2.3. Biosystems, 97:134–
139.
Osari, K., Murooka, T., Hagiwara, K., Ando, T., Shima-
kawa, M., Ito, S. , Hagihara, S., and Yonezaki, N.
(2014). An object-oriented language for parameteri-
sed reactive system specification based on li near tem-
poral logic. In Theory and Practice of Computation,
pages 121–143. WORLD SCIENTIFIC.
Palsson, B . (2000). The challenges of in silico biology. Nat.
Biotechnol., 18:1147–50.
Pnueli, A. and Rosner, R. (1989). On the synthesis of a re-
active module. In P OPL ’89: Proceedings of the 16th
ACM SIGPLAN-SIGACT symposium on Principles of
programming languages, pages 179–190, New York,
NY, USA. AC M.
Rizk, A., Batt, G., Fages, F., and Soliman, S. (2009). A
general computational method for robustness analysis
with applications to synthetic gene networks. Bioin-
formatics, 25(12):i169–i178.
Schaub, M. A., Henzinger, T. A., and Fisher, J. (2007). Qua-
litative networks: A symbolic approach to analyze bi-
ological signaling networks. BMC Systems Biology,
1:4.
Thomas, R. (1991). Regulatory networks seen as asynchro-
nous automata: A logical description. J. Theor. Biol.,
153(1):1–23.
Vanitha, V., Yamashita, K., Fukuzawa, K., and Yonezaki.,
N. (2000). A method for structuralisation of evolutio-
nal specifications of reactive systems. In ICSE 2000,
The Third International Workshop on Intelligent Soft-
ware Engineering (WISE3), pages 30 – 38.
Wang, Q., Z uliani, P., Kong, S., Gao, S., and Clarke,
E. M. (2015). SReach: A probabilistic bounded delta-
reachability analyzer for stochastic hybrid systems. In
Roux, O. and Bourdon, J., editors, CMSB 2015, vo-
lume 9308 of LNCS, pages 15–27, C ham. Springer
International Publishing.
Zhang, Q. and Andersen, M. E. (2007). Dose response rela-
tionship in anti-stress gene regulatory networks. PLoS
Comput. Biol., 3(3).
BIOINFORMATICS 2017 - 8th International Conference on Bioinformatics Models, Methods and Algorithms
28
