
Ab initio Splice Site Prediction with
Simple Domain Adaptation Classifiers
Nic Herndon and Doina Caragea
Computing and Information Sciences, Kansas State University, Manhattan, KS 66506, U.S.A.
Keywords:
Splice Site Prediction, Domain Adaptation, Imbalanced Data, Logistic Regression, Na
¨
ıve Bayes.
Abstract:
The next generation sequencing technologies (NGS) have made it affordable to sequence any organism, open-
ing the door to assembling new genomes and annotating them, even for non-model organisms. One option
for annotating a genome is to assemble RNA-Seq reads into a transcriptome and aligning the transcriptome to
the genome assembly to identify the protein-encoding genes. However, there are a couple of problems with
this approach. RNA-Seq is error prone and therefore the gene models generated with this technique need to
be validated. In addition, this method can only capture the genes expressed at the time of sequencing. Ma-
chine learning can help address both of these problems by generating ab initio gene models that can provide
supporting evidence to the models generated with RNA-Seq, as well as predict additional genes that were not
expressed during sequencing. However, machine learning algorithms need large amounts of labeled data to
learn accurate classifiers, and newly sequenced, non-model organisms have insufficient labeled data. This can
be addressed by leveraging the abundant labeled data from a related model-organism (the source domain) and
use it in conjunction with the little labeled data from the organism of interest (the target domain) to train a
classifier in a domain adaptation setting. The method we propose uses this approach and generates accurate
classification on the task of splice site prediction – a difficult and essential step in gene prediction. It is sim-
ple – it combines source and target labeled data, with different weights, into one dataset, and then trains a
supervised classifier on the combined dataset. Despite its simplicity it is surprisingly accurate, with highest
areas under the precision-recall curve between 53.33% and 83.57%. Out of the domain adaptation classifiers
evaluated (SVM, na
¨
ıve Bayes, and logistic regression) this method produced the best results in 12 out of the
16 cases studied.
1 INTRODUCTION
Recently a number of domain adaptation algorithms
have been proposed to address the lack of labeled data
in the domain of interest, the target domain, by lever-
aging plentiful labeled data from a related domain, the
source domain, and in some cases the large volume of
unlabeled data available from the target domain. One
application that meets this criteria – lacking labeled
data and with abundant labeled data in a similar do-
main – that is tackled by these algorithms is splice
site prediction. This was enabled by the next genera-
tion sequencing technologies, which allow faster and
cheaper sequencing of DNA and RNA than the previ-
ously used Sanger technology, leading to advances in
the field of genomics.
With NGS, short DNA read fragments are used to
generate genome assemblies. Similarly, RNA frag-
ments are assembled into transcriptomes. The tran-
scriptome is then used as evidence when annotating a
genome, by mapping it along that genome. This helps
determine the location and structure of the protein-
encoding genes. One of the disadvantages of this
method is that RNA-Seq reads are generated only
from the genes expressed at the time of sequencing in
the tissue analyzed, leaving out of the transcriptome
some of the protein-encoding genes.
In addition, assembling the transcriptome is not
error proof. NGS technologies speed up the sequenc-
ing of DNA and RNA molecules, but do so at the
expense of read length and accuracy. They gener-
ate shorter reads than previous sequencing technolo-
gies (e.g., Sanger) with much higher error rates. The
common practice to address these issues is to trim the
low quality ends of the reads, remove reads with low
scores, and require higher depth of coverage. The re-
maining reads are then assembled into a genome (for
DNA reads) or transcriptome (for RNA reads). These
assemblies are not 100% accurate. Therefore, anno-
tating a genome with a transcriptome generated from
Herndon, N. and Caragea, D.
Ab initio Splice Site Prediction with Simple Domain Adaptation Classifiers.
DOI: 10.5220/0005710502450252
In Proceedings of the 9th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2016) - Volume 3: BIOINFORMATICS, pages 245-252
ISBN: 978-989-758-170-0
Copyright
c
2016 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
245

RNA-Seq reads should be validated by independent
methods (Steijger et al., 2013).
Domain adaptation algorithms can provide a
means to validate the gene models produced with this
technique, as well as generate gene models for genes
missed by RNA-Seq. Such classifiers can accom-
plish this despite the difficult nature of the splice site
prediction problem – a highly imbalanced problem,
where only a small ratio of the GT and AG dimers
within a genome are splice sites (Sonnenburg et al.,
2007). These are the donor and acceptor canonical
splice sites, respectively. Even though this type of
problem is difficult for every type of classifier – su-
pervised or semi-supervised – not just domain adap-
tation, the existing algorithms, discussed in Section 2,
achieved good results.
In this work, we propose an algorithm that sur-
passes these results. It is a simple, yet surprisingly
accurate method, presented in detail in Section 3.1.
With this method we combine data from two organ-
isms into one dataset. Each organism is assigned a
different weight. The resulting dataset is then used
to train a supervised classifier for the organism of in-
terest. To evaluate this algorithm we tested it with
data from C.elegans, the source domain, and four tar-
get domains at increasing evolutionary distance from
this source: C.remanei, P.pacificus, D.melanogaster,
and A.thaliana – data described in Section 3.2. From
the results shown in Section 4, we can infer that this
method is a viable ab initio splice site prediction tech-
nique. It generated highest average areas under the
precision-recall curve (auPRC) for the positive class
between 53.33% for distantly related organisms and
83.57% for closely related ones.
2 RELATED WORK
Most of the research on ab initio splice prediction fo-
cused on supervised learning approaches. Most meth-
ods proposed used either support vector machines,
(Baten et al., 2006; Li et al., 2012; Sonnenburg et al.,
2007; Zhang et al., 2006), Bayesian networks, (Cai
et al., 2000), hidden Markov models, (Baten et al.,
2007), or Bahadur expansion truncated at the second
order, (Arita et al., 2002). However, as these methods
employ supervised classifiers, they generally require
lots of labeled data to generate accurate predictions.
Other methods evaluated the use of semi-
supervised classifiers for this task. One study in-
vestigated one of the main factors that affects the
performance of an expectation-maximization semi-
supervised algorithm – the highly imbalanced class
distribution (Stanescu and Caragea, 2014b). The au-
thors studied the effects of the level of imbalance
on the accuracy of the classifier, and recommended
different ways to address it: adding only instances
from the minority class at each iteration, balancing
the class ratio through oversampling, and splitting the
data into balanced subsets by undersampling and then
training an ensemble of classifiers on these datasets.
In their subsequent study (Stanescu and Caragea,
2014a), they further analyzed ensemble-based semi-
supervised learning approaches, and recommended
using an ensemble of self-training classifiers that add
at each iteration only instances from the minority
class. However, considering the highly imbalanced
nature of the problem and the lack of sufficient la-
beled data, the accuracy of these classifiers was not
very high, with the highest auPRC of 54.78% for the
best classifier.
For domain adaptation setting, there are several
studies. One proposed an iterative domain adaptation
algorithm derived from na
¨
ıve Bayes, that used source
data, and target labeled and unlabeled data (Herndon
and Caragea, 2014b). Although it performed well
on the task of protein localization, it produced un-
satisfactory results for splice site prediction. Their
first updated version (Herndon and Caragea, 2014a)
produced promising results for splice site prediction
with highest auPRC between 43.20% for distant do-
mains and 78.01% for related domains. Later, they
achieved even better results, with best auPRCs be-
tween 50.83% and 82.61%, (Herndon and Caragea,
2015). Another study for splice site prediction pro-
posed a modified version of the k-means cluster-
ing algorithm that considered the commonalities be-
tween the source and target domains (Giannoulis
et al., 2014). This algorithm was not very accurate
though. Its best area under receiver operating char-
acteristic curve (auROC) was below 70%. One of
the best methods for splice site prediction in this set-
ting, used a support vector machine classifier, with
highest auPRC values between 49.75% and 79.02%,
(Schweikert et al., 2009).
There are also evidence-based methods, such as
TWINSCAN (Korf et al., 2001), CONTRAST (Gross
et al., 2007), TrueSight (Li et al., 2013), and us-
ing single-molecule transcript sequencing (Minoche
et al., 2015). It is however unfair to compare these
with ab initio methods, as they use mRNA evidence
to generate their models, whereas ab initio methods
do not.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
246

3 METHOD AND MATERIALS
3.1 Proposed Method
Let the set of independently generated training in-
stances be represented by X ∈ R
m×n
and their corre-
sponding labels by y ∈ Y
m
, Y = {0, 1}, where m is
the number of training instances and n is the number
of features.
Given a set of training instances from the source
domain, D
S
= (X
S
, y
S
), where X
S
∈ R
m
S
×n
and y
S
∈
Y
m
S
, and a set of training instances from the tar-
get domain, D
T
= (X
T
, y
T
), where X
T
∈ R
m
T
×n
and
y
T
∈ Y
m
T
, create an empty dataset D = (X, y), where
X ∈ R
(m
S
+m
T
)×n
and y ∈ Y
m
S
+m
T
. For each instance
(x
i
, y
i
) ∈ D
S
multiply its weight by w
S
, then add
this instance to the new dataset, D. Similarly, for
each instance (x
j
, y
j
) ∈ D
T
multiply its weight by
w
T
, then add it to the new dataset, D. Then, train
a supervised classifier on this combined dataset, D.
In our experiments we used WEKA implementations
of the regularized logistic regression (Le Cessie and
Van Houwelingen, 1992), and na
¨
ıve Bayes (John and
Langley, 1995) classifiers.
3.2 Data Sets
We evaluated our proposed method using the same
dataset
1
as in the previous related domain adap-
tation studies, dataset that was first introduced in
(Schweikert et al., 2009). It contains DNA sequences
from one source organism, C.elegans, and four target
organisms at increasing evolutionary distance from
source, C.remanei, P.pacificus, D.melanogaster, and
A.thaliana. For the source organism there is one
dataset with 100,000 instances, and for each target or-
ganism there are three folds of 1,000, 2,500, 6,500,
16,000, 25,000, 40,000, and 100,000 instances used
for training, and three folds of 20,000 instances used
for testing. Each instance is a 141 nucleotides long
DNA sequence, with the AG dimer at the sixty-first
position, and the label for each instance indicates
whether this dimer is an acceptor splice site (positive)
or not (negative). In each file about 1% of instances
are positive and the remaining are negative.
3.3 Experimental Setup
From this dataset, to compare our proposed method
with previous methods, we used the three folds of
1
Downloaded from ftp://ftp.tuebingen.mpg.de/
fml/cwidmer/
2,500, 6,500, 16,000, and 40,000 instances as tar-
get labeled data. Note that the method proposed by
(Herndon and Caragea, 2014a), also uses the three
folds of 100,000 instances from the target organisms
as unlabeled data, which have the potential to increase
the accuracy of the classifier.
We use the same representation for the data as in
(Herndon and Caragea, 2014a; Herndon and Caragea,
2015). Namely, we convert the DNA sequences into
two types of features, nucleotides and trimers, along
with their position within the sequence. In one set
of experiments we represent the data with nucleotide
features only, and in the other we represent it with
both types of features. For example, using nucleotide
and trimer features, a DNA sequence starting with
TTCTAAGCG. . . and class 1 would be represented in
WEKA ARFF format as:
@RELATION rel
@ATTRIBUTE NUCLEOTIDE 1 {A,C,G,T}
.
.
.
@ATTRIBUTE NUCLEOTIDE 141 {A,C,G,T}
@ATTRIBUTE TRIMER 1 {AAA,AAC,. . .,TTT}
.
.
.
@ATTRIBUTE TRIMER 139 {AAA,AAC,. . .,TTT}
@ATTRIBUTE cls {1,-1}
@DATA
T,T,C,T,A,A,G,C,G,. . .,TTC,TCT,CTA,. . .,1
We would like to note that the trimer features are
not independent of each other. Each trimer has nu-
cleotides in common with the overlapping neighbor-
ing trimers – two to five neighbors, depending on the
position of the trimer. The trimers at each end of a se-
quence have nucleotides in common with two neigh-
boring trimers. The trimers in the middle, have nu-
cleotides in common with at most five neighbors. This
does not violate the independence assumption of the
na
¨
ıve Bayes classifiers. These classifiers still assume
that all features are independent of each other.
To find the optimal parameters’ values we did a
grid search for w
S
, w
T
∈ {0.1, 0.2, . . . , 1}, using the
target datasets of 100,000 instances for validation
(same as was done in the method proposed by (Hern-
don and Caragea, 2015)). For our proposed method
we:
1. Trained the classifier with labeled data from the
source and target domains.
2. Evaluated on the validation dataset and picked the
values for w
S
and w
T
that generated best auPRC.
3. Tested the classifier with these parameters’ values
on the target domain.
For the source domain we used the only dataset, with
Ab initio Splice Site Prediction with Simple Domain Adaptation Classifiers
247

100,000 instances. For the target domain, for each
organism we used:
• For training, one of the three folds of 2,500, 6,500,
16,000 or 40,000 instances.
• For validation, the corresponding fold of 100,000
instances.
• For testing, the corresponding fold of 20,000 in-
stances.
As baselines, we used the na
¨
ıve Bayes and the
logistic regression with regularized parameters clas-
sifiers, trained on either 100,000 from C.elegans, or
one of the three folds of 2,500, 6,500, 16,000, or
40,000 from the target organisms, and tested them
on the corresponding fold for that organism. We ex-
pect the results of the baseline classifiers will be the
lower bound for our proposed method, as we hypoth-
esize that adding data from a related organism should
improve the accuracy of the classifier. Note, that
whenever we used the logistic regression classifier,
for baselines or for our proposed method, we set the
ridge parameter to 1,000, as this value led to the best
results in (Herndon and Caragea, 2015).
All results are reported in Table 1 as averages of
three random train-test splits, to ensure the results are
not biased. For evaluating the classifiers we used the
area under the precision-recall curve for the minority
class, which is the class of interest, since the data are
so highly imbalanced (Davis and Goadrich, 2006).
This experimental setup allowed us to evaluate:
1. How the following factors influence the perfor-
mance of our classifier: features, amount of tar-
get labeled data, distance between domains, and
weights used for source and target data.
2. How our proposed method (when using na
¨
ıve
Bayes or regularized logistic regression) com-
pares to other domain adaptation classifiers for the
task of splice site prediction, namely, the SVM
classifier proposed by (Schweikert et al., 2009),
the na
¨
ıve Bayes classifier proposed by (Herndon
and Caragea, 2014a), and the regularized logis-
tic regression proposed by (Herndon and Caragea,
2015).
4 RESULTS AND DISCUSSION
In Table 1 we show the auPRC averages over three
folds and their standard deviations for the four target
organisms for:
• Our proposed method (LR SL
S+T
when using the
regularized logistic regression and NB SL
S+T
when
using the na
¨
ıve Bayes classifier).
• Supervised classifiers used as baselines (LR SL
S
and LR SL
T
when using the regularized logistic
regression classifier trained on source and target
data, respectively, and NB SL
S
and NB SL
T
when
using the na
¨
ıve Bayes classifier trained on source
and target data, respectively).
• The domain adaptation with na
¨
ıve Bayes classi-
fier proposed by (Herndon and Caragea, 2014a)
(NB DA
S+T+U
). Note that this is the only classifier,
from the ones we compared, that used the target
unlabeled data in addition to the source and target
labeled data.
• The domain adaptation with regularized logistic
regression proposed by (Herndon and Caragea,
2015) (LR cc).
• The domain adaptation with SVM classifier pro-
posed by (Schweikert et al., 2009) (SVM). Note
that this classifier used other features to represent
the DNA sequences (i.e., it did not represent them
with nucleotides and trimers along with their po-
sitions).
Based on these results we make the following ob-
servations:
1. In terms of the different factors that influence the
performance of the classifier:
(a) Features: we notice a similar trend for our
proposed method as with previous classifiers
(Herndon and Caragea, 2014a; Herndon and
Caragea, 2015), namely, using simple features
(the nucleotides) leads to more accurate clas-
sifiers when the source and target domains are
distant and there is scarce labeled data in the
target domain. Using a combination of simple
and complex features (nucleotides and trimers)
leads to more accurate classifiers when the
source and target domains are closed and there
is enough target labeled data. This is expected
as trimer features are sparser than nucleotide
features, and with less labeled data the classi-
fier performs worse with trimer features as it
does not have enough data to learn an accurate
classifier.
(b) Amount of Target Labeled Data: as the
amount of target labeled data increases the
accuracy of our proposed method increases
as well, with one exception, though. For
D.melanogaster, when using nucleotide and
trimer features, we observe the auPRC de-
creases as the amount of target labeled data in-
creases from 16,000 to 40,000, regardless of
the type of supervised classifier we used, na
¨
ıve
Bayes, or regularized logistic regression. It is
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
248
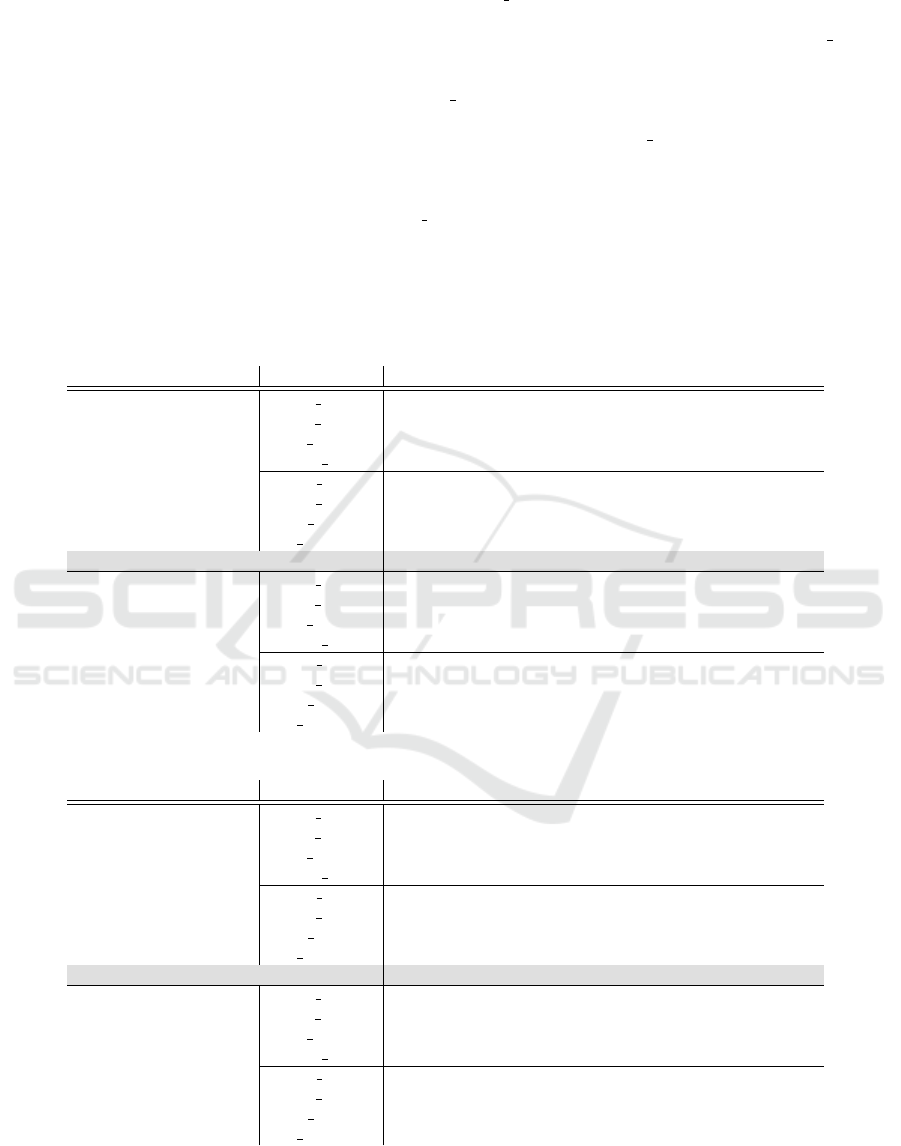
Table 1: auPRC values for the minority (i.e., positive) class for four target organisms based on the number of labeled target
instances used for training: 2,500, 6,500, 16,000, and 40,000. The LR SL classifier is the logistic regression classifier trained
on 100,000 instances from the source domain, C.elegans (first and tenth rows); trained on target labeled data (second and
eleventh rows); and a combination of source and target labeled data (third and twelfth rows), respectively. LR cc (rows
forth and thirteenth) is the domain adaptation classifier trained on a combination of source labeled and target labeled data in
(Herndon and Caragea, 2015). SVM (ninth rows) is the best overall classifier in (Schweikert et al., 2009), namely SVM
S,T
.
Note that the SVM classifier used different features. The NB SL classifier is the na
¨
ıve Bayes classifier trained on 100,000
instances from the source domain (fifth and thirteenth rows); trained on target labeled data (sixth and fourteenth rows); and a
combination of source and target labeled data (seventh and fifteenth rows), respectively. NB DA
S+T+U
is the best overall domain
adaptation classifier in (Herndon and Caragea, 2014a), A1, trained on a combination of source labeled, target labeled, and
target unlabeled data. The best average values for each type of features used is shown in bold font. (Note that for P.pacificus
the best auPRC value when using nucleotides and trimers with 2,500 and 6,500 target labeled instances is 67.10, obtained
with the na
¨
ıve Bayes classifier trained on source data, NB
SL
S
.) We would like to highlight that when the source and target
domains are close (C.remanei and P.pacificus are close to C.elegans), the best overall classifier is logistic regression trained
on the combination of source labeled and target labeled data (i.e., best auPRC values in six out of eight cases). When the
source and target domains are distant (D.melanogaster and A.thaliana are far from C.elegans), the best overall classifier is
na
¨
ıve Bayes trained on the combination of source labeled and target labeled data (i.e., best auPRC values in five out of eight
cases).
(a) C.remanei
Features Classifier 2,500 6,500 16,000 40,000
nucleotides
LR SL
S
77.63±1.37
LR SL
T
31.07±8.72 54.20±3.97 65.73±2.76 72.93±1.70
LR SL
S+T
77.65±1.34 77.88±1.16 78.32±1.29 79.00±0.97
LR cc 77.64±1.39 77.75±1.25 77.88±1.42 78.10±1.15
NB SL
S
63.77±1.30
NB SL
T
23.42±7.39 45.44±4.01 54.57±2.63 59.68±1.62
NB SL
S+T
75.49±1.39 75.56±1.46 75.63±1.45 75.82±1.32
NB DA
S+T+U
59.18±1.17 63.10±1.23 63.95±2.08 63.80±1.41
SVM 77.06±2.13 77.80±2.89 77.89±0.29 79.02±0.09
nucleotides and trimers
LR SL
S
81.37±2.27
LR SL
T
26.93±9.91 55.26±2.21 68.30±1.91 77.33±2.78
LR SL
S+T
81.40±2.25 81.73±1.90 82.62±2.28 83.57±1.76
LR cc 81.39±2.30 81.47±2.19 81.78±2.08 82.61±2.00
NB SL
S
77.67±2.24
NB SL
T
22.94±4.37 58.39±3.94 68.40±3.37 75.75±1.32
NB SL
S+T
81.11±0.73 81.38±0.34 81.51±0.87 82.73±0.52
NB DA
S+T+U
45.29±2.62 72.00±4.16 74.83±4.32 77.07±4.45
(b) P.pacificus
Features Classifier 2,500 6,500 16,000 40,000
nucleotides
LR SL
S
64.20±1.91
LR SL
T
29.87±3.58 49.03±4.90 59.93±2.74 69.10±2.25
LR SL
S+T
64.72±1.85 65.63±1.82 67.09±1.29 70.76±2.08
LR cc 64.70±1.85 65.31±2.10 66.76±0.89 70.18±2.12
NB SL
S
49.12±1.58
NB SL
T
19.22±3.39 37.33±2.65 45.33±2.28 52.84±2.06
NB SL
S+T
60.67±1.97 61.96±2.04 63.04±0.33 65.17±2.09
NB DA
S+T+U
45.32±2.68 49.82±2.58 52.09±2.04 54.62±1.51
SVM 64.72±3.75 66.39±0.66 68.44±0.67 71.00±0.38
nucleotides and trimers
LR SL
S
62.37±0.84
LR SL
T
28.40±4.49 49.67±2.83 62.97±3.32 74.60±2.85
LR SL
S+T
64.14±0.83 66.14±0.55 70.97±2.03 76.89±1.75
LR cc 64.18±1.10 65.49±1.84 69.76±2.08 75.82±2.00
NB SL
S
67.10±1.94
NB SL
T
26.39±3.97 48.54±3.42 59.29±2.80 68.78±1.52
NB SL
S+T
64.51±0.70 66.32±0.71 69.29±2.00 72.54±0.42
NB DA
S+T+U
20.21±1.17 53.29±3.08 62.33±3.60 69.88±4.04
Ab initio Splice Site Prediction with Simple Domain Adaptation Classifiers
249

Table 1: (Continued)
(c) D.melanogaster
Features Classifier 2,500 6,500 16,000 40,000
nucleotides
LR SL
S
35.87±2.32
LR SL
T
19.97±3.48 31.80±3.86 42.37±2.15 50.53±1.80
LR SL
S+T
41.35±1.40 43.66±3.20 49.96±2.09 54.02±0.95
LR cc 39.70±2.82 42.19±3.41 49.72±2.01 53.43±0.89
NB SL
S
31.23±1.03
NB SL
T
14.90±2.80 26.05±4.79 35.21±2.43 39.42±2.90
NB SL
S+T
45.43±0.87 47.12±3.86 51.73±1.24 52.74±2.43
NB DA
S+T+U
33.31±3.71 36.43±2.18 40.32±2.04 42.37±1.51
SVM 40.80±2.18 37.87±3.77 52.33±0.91 58.17±1.50
nucleotides and trimers
LR SL
S
32.23±2.76
LR SL
T
15.07±4.11 28.30±5.45 44.67±3.23 38.43±32.36
LR SL
S+T
34.97±2.59 37.22±4.30 49.16±5.11 43.03±22.03
LR cc 37.24±2.20 40.93±3.79 50.54±3.91 45.89±22.25
NB SL
S
34.09±2.44
NB SL
T
13.87±2.97 25.00±5.59 35.28±2.14 45.85±3.32
NB SL
S+T
46.85±1.41 50.84±4.39 56.57±2.37 50.15±14.84
NB DA
S+T+U
25.83±2.35 32.58±5.83 39.10±1.82 47.49±3.44
(b) A.thaliana
Features Classifier 2,500 6,500 16,000 40,000
nucleotides
LR SL
S
16.93±0.21
LR SL
T
13.87±2.63 26.03±3.29 38.43±6.18 49.33±4.07
LR SL
S+T
22.79±0.92 31.70±2.70 41.28±2.64 49.91±2.38
LR cc 20.67±0.58 27.19±1.30 40.56±3.26 49.75±2.82
NB SL
S
11.97±0.23
NB SL
T
7.21±0.90 17.90±1.93 28.10±4.68 34.82±4.77
NB SL
S+T
23.30±1.18 30.97±2.31 39.18±2.79 44.88±3.13
NB DA
S+T+U
18.46±1.13 25.04±0.72 31.47±3.56 36.95±3.39
SVM 24.21±3.41 27.30±1.46 38.49±1.59 49.75±1.46
nucleotides and trimers
LR SL
S
14.07±0.31
LR SL
T
8.87±1.84 21.10±4.45 38.53±8.08 49.77±2.77
LR SL
S+T
15.87±0.36 23.65±1.49 39.97±4.39 50.60±2.11
LR cc 16.42±1.20 26.44±2.49 41.35±6.49 50.83±2.28
NB SL
S
13.98±0.71
NB SL
T
3.10±0.35 8.76±1.65 28.21±7.58 40.92±3.78
NB SL
S+T
21.62±1.02 27.89±2.19 43.52±6.16 53.33±3.77
NB DA
S+T+U
3.99±0.43 13.96±2.42 33.62±6.31 43.20±3.78
interesting to note that for this combination of
features used and target domain, the auPRC
for the regularized logistic regression classi-
fier also decreases when the amount of target
labeled data increases from 16,000 to 40,000.
This partially explains this exception for our
proposed method when using the logistic re-
gression classifier. Another factor, suggested
by the large standard deviation, is that the fre-
quency of features is very different between
training and test datasets, especially for trimers,
since using only nucleotide features does not
exhibit this behavior.
(c) Distance between Domains: as the distance
between the source and target domains in-
creases, the contribution from the source data
decreases, and the accuracy of our method de-
creases, which is expected.
(d) Weight Assigned to Source and Target Data:
in regards to the weight assigned to the target
labeled data, the best results are obtained when
w
T
is set to one, or close to one. For the weight
assigned to the source labeled data, when the
domains are closely related the best results are
for high values of w
S
, but as the distance be-
tween domains increases the value for w
S
de-
creases. It only makes sense to decrease the
weight assigned to source data when the dis-
tance between domains increases, so these re-
sults confirm our intuition.
2. In terms of performance, our proposed method
produced the best results out of all domain adap-
tation classifiers compared, when the source and
target domains are closely related (for C.remanei
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
250

and P.pacificus)), using logistic regression with
nucleotide and trimer features. It also produced
the best results when the domains are distant
(for D.melanogaster and A.thaliana), using na
¨
ıve
Bayes with nucleotide and trimer features, in five
out of eight cases. This is a similar behavior to the
one observed in (Ng and Jordan, 2001), namely
that a generative classifier performs better than a
discriminative one when there is a small amount
of training labeled data. For domain adaptation,
when the domains are close the source labeled
data contributes a lot to the classifier so a discrim-
inative classifier performs better than a generative
one. When the domains are distant, the source la-
beled data contributes less and a generative classi-
fier performs better than a discriminative one. An-
other case for which our method produced the best
results is for very distant domains (A.thaliana),
using logistic regression with nucleotide features,
when there is somewhat scarce target labeled data
(6,500 instances). There are only two cases in
which another domain adaptation classifier, the
SVM proposed by (Schweikert et al., 2009), out-
performed our proposed method.
5 CONCLUSIONS AND FUTURE
WORK
In this paper we proposed a simple domain adapta-
tion method to address the lack of or limited amount
of labeled data for a target domain, by leveraging the
large amount of labeled data from a related domain.
We evaluated this method on a biological problem,
splice site prediction, a critical step for gene annota-
tion, since many organisms have limited to no labeled
data, whereas related, more studied model organisms
have large amounts of labeled data.
From our experimental results we made a few ob-
servations, such as, in some cases simple features are
preferred over complex ones when the latter can lead
to sparse representations and decreased accuracy, and
vice versa; using more labeled data increases the ac-
curacy of the classifier; and that as the distance be-
tween the domains increases the contribution of the
source data decreases. More importantly, we ob-
served that our proposed method performed better
than previously proposed methods with only a cou-
ple of exceptions, recommending it for ab initio splice
site prediction.
For future work we would like to explore ways to
further increase its accuracy. For example, we would
like to create balanced subsamples, through under-
sampling, and then training an ensemble of classifiers
on these subsamples. In addition, we would like to
experiment with ensembles of classifiers produced by
the different methods proposed, on balanced datasets.
Another direction for future work is to combine data
from multiple organisms and train a classifier for a
target organism, i.e., use multiple source domains.
ACKNOWLEDGEMENTS
This work was supported by an Institutional Devel-
opment Award (IDeA) from the National Institute of
General Medical Sciences of the National Institutes
of Health under grant number P20GM103418. The
content is solely the responsibility of the authors and
does not necessarily represent the official views of
the National Institute of General Medical Sciences or
the National Institutes of Health. The computing for
this project was performed on the Beocat Research
Cluster at Kansas State University, which is funded
in part by grants MRI-1126709, CC-NIE-1341026,
MRI-1429316, CC-IIE-1440548.
REFERENCES
Arita, M., Tsuda, K., and Asai, K. (2002). Modeling splic-
ing sites with pairwise correlations. Bioinformatics,
18(suppl 2):S27–S34.
Baten, A. K., Chang, B. C., Halgamuge, S. K., and Li, J.
(2006). Splice site identification using probabilistic
parameters and svm classification. BMC bioinformat-
ics, 7(Suppl 5):S15.
Baten, A. K., Halgamuge, S. K., Chang, B., and Wickrama-
rachchi, N. (2007). Biological sequence data prepro-
cessing for classification: A case study in splice site
identification. In Advances in Neural Networks–ISNN
2007, pages 1221–1230. Springer.
Cai, D., Delcher, A., Kao, B., and Kasif, S. (2000). Model-
ing splice sites with bayes networks. Bioinformatics,
16(2):152–158.
Davis, J. and Goadrich, M. (2006). The relationship be-
tween precision-recall and roc curves. In Proceed-
ings of the 23rd international conference on Machine
learning, pages 233–240. ACM.
Giannoulis, G., Krithara, A., Karatsalos, C., and Paliouras,
G. (2014). Splice site recognition using transfer learn-
ing. In SETN, pages 341–353. Springer.
Gross, S. S., Do, C. B., Sirota, M., and Batzoglou, S.
(2007). Contrast: a discriminative, phylogeny-free ap-
proach to multiple informant de novo gene prediction.
Genome biology, 8(12):R269.
Herndon, N. and Caragea, D. (2014a). Empirical Study of
Domain Adaptation Algorithms on the Task of Splice
Site Prediction. Communications in Computer and In-
formation Science (CCIS 2014). Springer-Verlag.
Ab initio Splice Site Prediction with Simple Domain Adaptation Classifiers
251

Herndon, N. and Caragea, D. (2014b). Predicting pro-
tein localization using a domain adaptation approach.
In Biomedical Engineering Systems and Technologies,
pages 191–206. Springer.
Herndon, N. and Caragea, D. (2015). Domain adaptation
with logistic regression for the task of splice site pre-
diction. In 11th International Symposium on Bioin-
formatics Research and Applications, ISBRA 2015,
pages 125–137.
John, G. H. and Langley, P. (1995). Estimating continuous
distributions in bayesian classifiers. In Proceedings
of the Eleventh conference on Uncertainty in artificial
intelligence, pages 338–345. Morgan Kaufmann Pub-
lishers Inc.
Korf, I., Flicek, P., Duan, D., and Brent, M. R. (2001). Inte-
grating genomic homology into gene structure predic-
tion. Bioinformatics, 17(suppl 1):S140–S148.
Le Cessie, S. and Van Houwelingen, J. C. (1992). Ridge
estimators in logistic regression. Applied statistics,
pages 191–201.
Li, J., Wang, L., Wang, H., Bai, L., and Yuan, Z. (2012).
High-accuracy splice site prediction based on se-
quence component and position features. Genet Mol
Res, 11(3):3431–3451.
Li, Y., Li-Byarlay, H., Burns, P., Borodovsky, M., Robin-
son, G. E., and Ma, J. (2013). Truesight: a new algo-
rithm for splice junction detection using rna-seq. Nu-
cleic acids research, 41(4):e51–e51.
Minoche, A. E., Dohm, J. C., Schneider, J., Holtgr
¨
awe, D.,
Vieh
¨
over, P., Montfort, M., S
¨
orensen, T. R., Weis-
shaar, B., and Himmelbauer, H. (2015). Exploiting
single-molecule transcript sequencing for eukaryotic
gene prediction. Genome biology, 16(1):1–13.
Ng, A. Y. and Jordan, M. I. (2001). On discriminative
vs. generative classifiers: a comparison of logistic
regression and naive bayes. In Proceedings of the
Neural Information Processing Systems Conference,
pages 841–848.
Schweikert, G., R
¨
atsch, G., Widmer, C., and Sch
¨
olkopf,
B. (2009). An empirical analysis of domain adap-
tation algorithms for genomic sequence analysis. In
Advances in Neural Information Processing Systems,
pages 1433–1440.
Sonnenburg, S., Schweikert, G., Philips, P., Behr, J., and
R
¨
atsch, G. (2007). Accurate splice site prediction us-
ing support vector machines. BMC bioinformatics,
8(Suppl 10):S7.
Stanescu, A. and Caragea, D. (2014a). Ensemble-
based semi-supervised learning approaches for im-
balanced splice site datasets. In Bioinformatics and
Biomedicine (BIBM), 2014 IEEE International Con-
ference on, pages 432–437. IEEE.
Stanescu, A. and Caragea, D. (2014b). Semi-supervised
self-training approaches for imbalanced splice site
datasets. In Proceedings of the 6th International Con-
ference on Bioinformatics and Computational Biol-
ogy, BICoB, pages 131–136.
Steijger, T., Abril, J. F., Engstr
¨
om, P. G., Kokocinski, F.,
Hubbard, T. J., Guig
´
o, R., Harrow, J., Bertone, P.,
Consortium, R., et al. (2013). Assessment of transcript
reconstruction methods for rna-seq. Nature methods,
10(12):1177–1184.
Zhang, Y., Chu, C.-H., Chen, Y., Zha, H., and Ji, X. (2006).
Splice site prediction using support vector machines
with a bayes kernel. Expert Systems with Applications,
30(1):73–81.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
252
