
Temporal Logic based Framework to Model and Analyse Gene Networks
with Alternative Splicing
Sohei Ito
Department of Fisheries Distribution and Management, National Fisheries University, 2-7-1 Nagata-Honmachi,
Shimonoseki, Yamaguchi, Japan
Keywords:
Gene Regulatory Network, Systems Biology, Alternative Splicing, Temporal Logic.
Abstract:
Toward system-level understanding of biological systems, we need a formalism to model and analyse them.
Due to incompleteness of knowledge about quantitative parameters and molecular mechanisms, qualitative
methods have been useful alternatives. We have been working on temporal logic-based approach for qual-
itative modelling and analysis of gene regulatory networks. Although our framework is well-established to
model several aspects of gene regulation, we still lack treatment of alternative splicing, which contributes to
proteomic diversity of eukaryotic organisms. In this paper we extend our logic-based qualitative framework
to be able to capture alternative splicing, which is crucial to model the gene regulatory networks in eukaryotic
organisms. We study mechanisms of alternative splicing and propose how we model each mechanism, then
demonstrate the modelling method by analysing the regulatory network of sex determination in Drosophila
and verify that the network ensures sex determination.
1 INTRODUCTION
To understand complex activities of the cell, mathe-
matical and computational approach is indispensable.
For precise mathematical modelling, we need huge
amount of quantitative information. Such quantita-
tive information available, however, is unfortunately
limited and not sufficient despite of recent advances
in biology. Instead, a lot of qualitative information
about biological systems has been accumulated such
as schematic network representations of gene-gene
interactions, protein-protein interactions, signalling
pathways, and so on. Thus a qualitative method for
modelling and analysing biological processes based
on qualitative information is desired.
In this context, several computational formalisms
in biological modelling have been proposed: Boolean
network (Thomas, 1991), Petri net (Heiner et al.,
2008), timed automata (Batt et al., 2007) and process
algebra (Ciocchetta and Hillston, 2009), though all
of them are not necessarily qualitative. In these for-
malisms, the possible behaviours of a system can be
characterised by the traces of the model. Such com-
putational formalisms need concrete information on
molecular mechanisms and regulatory logics to con-
struct a model. Since biological information is in-
herently incomplete, it is pointed out that constraint-
based modelling is well-suited in biological mod-
elling (Palsson, 2000) in which we give several con-
straints reflecting incomplete knowledge on the sys-
tem to limit possible behaviours (solution space) of
biological systems.
In accordance with the motivation of constraint-
based modelling, we have been working on a logic-
based qualitative approach to model and analyse be-
haviours of gene regulatory networks (Ito et al., 2010;
Ito et al., 2013b; Ito et al., 2013a; Ito et al., 2014; Ito
et al., 2015) which uses linear temporal logic (LTL)
as the modelling language. In contrast to the orig-
inal constraint-based modelling paradigm which in-
tends to limit the quantitativepossible behaviours, our
approach aims to characterise qualitative possible be-
haviours using qualitative information of gene-gene
interactions which is represented as gene regulatory
networks. Since we only use qualitative information,
the reasoning is also limited to qualitative properties.
However, we can still analyse important properties of
gene networks such as oscillation, stability and reach-
ability, as it is pointed out that the overall behaviour
is relatively insensitive to the exact numerical values
of the kinetic constants (Palsson, 2000).
One of the difficulties in modelling and analysing
gene networks is the alternative splicing in eukary-
otic organisms. Alternative splicing of a precursor
mRNA (pre-mRNA) gives rise to multiple transcrip-
Ito, S.
Temporal Logic based Framework to Model and Analyse Gene Networks with Alternative Splicing.
DOI: 10.5220/0005655001510158
In Proceedings of the 9th International Joint Conference on Biomedical Engineering Systems and Technologies (BIOSTEC 2016) - Volume 3: BIOINFORMATICS, pages 151-158
ISBN: 978-989-758-170-0
Copyright
c
2016 by SCITEPRESS – Science and Technology Publications, Lda. All rights reserved
151

tion products from one gene. The selection of alter-
native splicing at an appropriate timing is critical for
cell differentiationand sex determination. In quantita-
tive approach, the splicing process can be modelled as
thermodynamical reactions (Louis et al., 2003; Wen,
2013). However, it is unclear how alternative splicing
is modelled in qualitative approach. The aim of this
paper is to establish a method for modelling alterna-
tive splicing in our LTL-based framework. In this pa-
per we study mechanisms of alternative splicing and
show how each mechanism can be modelled in LTL.
We demonstrate our formal framework by modelling
and analysing the network of sex determination in
Drosophila (Camara et al., 2008; Salz and Erickson,
2010).
The rest of this paper is organised as follows. In
section 2 we review our LTL-based framework for
modelling and analysing gene networks using LTL as
a baseline of this work. In section 3 we study mecha-
nisms of alternative splicing and present how we for-
mally model them in our framework. In section 4 we
demonstrate our formal framework in analysing the
network of sex determination in Drosophila. The fi-
nal section offers conclusion and future directions.
2 QUALITATIVE FRAMEWORK
FOR MODELLING AND
ANALYSING GENE
NETWORKS USING LTL
A gene network is represented as a directed graph
whose nodes and edges (labelled by +/-) represent
genes and regulation relation (activation/inhibition)
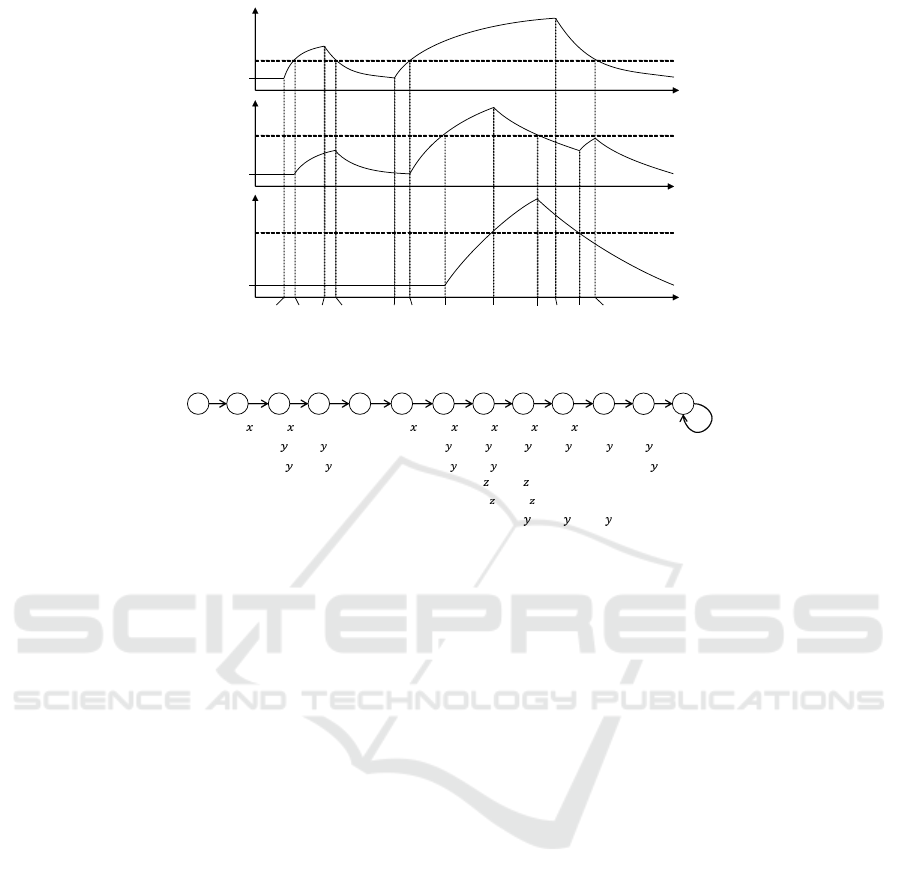
among them, respectively. In Fig. 1 we show an ex-
ample of a gene regulatory network which consists of
three genes. A behaviour of a gene network is rep-
resented as a time series of expression profiles of the
genes in the network. Fig. 2 shows an example time
series of the network depicted in Fig. 1. In this be-
haviour the value x
y
is the threshold level of gene x
to activate gene y, y
z
the threshold level of gene y to
activate gene z and z
y
the threshold level of gene z
to inhibit gene y. At the beginning, no genes are ex-
pressed. At time t
0
, gene x begins to be expressed
and its expression level begins to grow. At time t
1
gene x crosses the threshold x
y
, thus gene y becomes
ON due to the positive effect from gene x. At time
t
2
gene x stops to be expressed and the level decreas-
ing. At time t
3
gene x falls below x
y
, thus gene y
becomes OFF. This way the network changes its state
over time.
This time series can be represented as a discrete
y z
+
-
+
x
Figure 1: An example gene regulatory network. Gene x
activates gene y, y activates z and z inhibits y. A plus edge
represents activation and a minus edge represent inhibition.
state transition system (called linear time structure)
depicted in Fig. 3. It consists of states (represented as
circles) and transitions (represented as arrows). The
configurations of the network at each state are shown
by the propositions depicted below states. We have
the following propositions to describe the configura-
tions of the network:
• on
x
, on
y
, on
z
: whether genes x, y and z are ON or
OFF, respectively.
• x
y
, y
z
, z
y
: whether the expression level of gene x,
y and z are beyond the threshold x
y
, y
z
and z
y
,
respectively
1
.
We easily see that state 0 represents the configuration
of the network at the beginning, state 1 represents the
configuration betweent
0
and t
1
, state 2 between t
1
and
t
2
, and so on.
In general, there are many behaviours which can
be produced by a single network depending on the ini-
tial conditions, input scenarios, response times, and so
on. Our purpose is to model (or characterise) the set
of possible behaviours for a given gene network. In
quantitative approach, ordinary differential equations
(ODEs) are widely used. In the current setting, we
do not handle a numerical time series but a symbolic
time series of a behaviour (a linear time structure).
To characterise and reason about such structures, lin-
ear temporal logic (LTL) is the suitable mathemati-
cal language. LTL can be seen as propositional logic
equipped with temporal operators such as G (Glob-
ally), F (Future), U (Until) and W (Weak until). Gφ
means φ is always true, Fφ means φ is eventually true,
φUψ means φ is true until ψ is true and φWψ means
φ is true until ψ is true or φ is indefinitely true (in
this case ψ need not be true in any future). The for-
mal syntax and semantics are omitted due to the space
limitation.
We are to characterise the set of possible be-
haviours (linear time structures) for a given network.
This can be done by specifying an LTL formula φ
N
for a given network N such that the set of possible
behaviours of a network is characterised as {σ | σ |=
φ
N
}, i.e. all linear time structures which satisfy the
behaviour specification φ
N
. The problem of analysing
1
Threshold values are written in Roman while proposi-
tions are written in italics.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
152
x
y
z
t
base
base
base
0
1
2
3
4
5
6
7
8
9
x
୷
y
t
t
z
୷
1
0
1
1
Figure 2: Time series of expression levels of gene a, b and c in the network Fig. 1.
0
݊
݊
ݔ
݊
ݔ
݊
݊
݊
ݔ
݊
݊
ݔ
݊
ݕ
݊
ݔ
ݕ
݊
1 2 3 4 5 6 7 8 9 10
݊
ݖ
ݔ
݊
ݖ
11 12
ݔ
ݖ
݊
ݔ
Figure 3: Symbolic representation of the time series of Fig. 2.
network behaviours, e.g. checking whether there is a
behaviour which satisfies a certain property ψ (also
written in LTL), can be solved by finding σ such that
σ |= φ
N
∧ ψ, i.e. checking satisfiability of the for-
mula φ
N
∧ ψ. Thus analysing a gene network is re-
duced to satisfiability checking of LTL. Once we have
a formula φ
N
∧ ψ, the analysis can be automatically
done by LTL satisfiability checkers. LTL satisfiability
checkers construct a B¨uchi automaton of a given LTL
formula which precisely accepts the linear time struc-
tures in which the formula is true (Vardi and Wolper,
1994). Hence if the language accepted by the au-
tomaton is empty, the formula is not satisfiable. The
non-emptiness problem of B¨uchi automata is solved
by checking the existence of a maximal strongly con-
nected component containing accepting states of the
automata.
Due to the space limitation, we do not show in
detail how to specify φ
N
which characterises possible
behaviours of a given network N. Interested readers
would like to consult our previous work (Ito et al.,
2015). The key idea is the following qualitative prin-
ciples of gene network behaviours:
• A gene is ON when its activators are expressed
beyond some thresholds.
• A gene is OFF when its inhibitors are expressed
beyond some thresholds.
• If a gene is ON, its expression level increases.
• If a gene is OFF, its expression level decreases.
By expressing these principles in LTL, we have a
characterisation of possible behaviours of the net-
work. We only show an example characterisation
of the possible behaviours of the network in Fig. 1.
For this network we introduce the set of propositions
{on
x
, on
y
, on
z
, x
y
, y
z
, z
y
}. Using these propositions,
we have the following behaviour specification.
G(x
y
∧ ¬z
y
→ on
y
) ∧ G(z
y
→ ¬on
y
) ∧ G(y
z
↔ on
z
)∧
G(on
x
→ F(x
y
∨ ¬on
x
))∧
G(on
x
∧ x
y
→ (x
y
W¬on
x
))∧
G(¬on
x
→ F(¬x
y
∨ on
x
))∧
G(¬on
x
∧ ¬x
y
→ (¬x
y
Won
x
)) ∧ . . . .
In this specification we assumed that gene y is OFF if
gene z is inhibiting y nevertheless gene x is activating
y. For this network let us check the bistability of the
expression of gene y, which is written in LTL as:
(Gon
x
→ FGon
y
) ∧ (G¬on
x
→ FG¬on
y
)
We check the satisfiability of the conjunction of
the above formulae. We used T
3
-builder (Aoshima,
2003) to check it and had the answer ‘Yes’. This
means that the network of Fig. 1 produces two op-
posite behaviours: a behaviour in which gene y is al-
ways ON after some time point and another behaviour
in which gene y is always OFF after some time point,
which is determined by whether gene x is ON.
Temporal Logic based Framework to Model and Analyse Gene Networks with Alternative Splicing
153

3 MODELLING ALTERNATIVE
SPLICING BY LTL
In the formal framework described in the previous
section, we did not take alternative splicing into con-
sideration. This section discusses how we model the
alternative splicing in our framework.
In most eukaryotic organisms, the process of gene
expression consists of three steps: (i) a DNA region
which encodes a gene is transcribed into a precursor
messenger RNA (pre-mRNA), (ii) introns (and some
exons) in a pre-mRNA are removed, and (iii) a pro-
cessed mRNA is transported outside of a nucleus and
translated into a protein. Alternative splicing happens
in the step (ii) which causes the diversity of processed
mRNA from a single pre-mRNA by removing some
exons selectively as well as introns (Fig. 4). Due to
alternative splicing several isoforms of a protein are
obtained from one gene.
A natural solution to handle alternative splicing is
that we regard each isoform as being produced by dif-
ferent (virtual) genes. However, this treatment causes
blow-up of the number of propositions and the size of
a behaviour specification, which deteriorates the per-
formance of analysis.
In this section we study the molecular mech-
anisms of alternative splicing (David and Manley,
2008; Hertel, 2008; Kornblihtt, 2005; Matlin et al.,
2005) and propose how to model these mechanisms
in LTL without introducing extra genes.
Mechanism 1. One of the mechanisms of alterna-
tive splicing is the usage of multiple promoters. The
mouse α-amylase gene is known to have this mech-
anism. For the purpose of illustration, let us con-
sider a gene u which has two promoters X and Y (Fig.
5). Gene u has two splicing patterns depending on
promoters. The choice of promoters is made by a
transcription complex. To model this mechanism in
LTL, we introduce propositions TC
X
u
and TC
Y
u
to rep-
resent whether the levels of transcription complexes
for promoter X and Y are sufficient, respectively. We
write R
+
(u) and R
−
(u) for LTL terms representing
conditions for activation(+) and inhibition(-) of gene
u, respectively
2
. For example if gene v activates u
and gene w inhibits u, R
+
(u) will be v
u
∧ ¬w
u
and
R
−
(u) will be ¬v
u
∧ w
u
. Then conditions for activat-
ing/inhibiting gene u can be described as:
G(R
+
(u) ∧ TC
X
u
∧ ¬TC
Y
u
→ on
X
u
∧ ¬on
Y
u
), (1)
G(R
+
(u) ∧ TC
Y
u
∧ ¬TC
X
u
→ on
Y
u
∧ ¬on
X
u
), (2)
G(R
−
(u) → ¬on
X
u
∧ ¬on
Y
u
), (3)
2
In general we have several conditions for activat-
ing/inhibiting gene u, but treatment is the same.
where the propositions on
X
u
and on
Y
u
represent gene
u is expressed from promoter X and Y, respectively.
Formula (1) says that if gene u is activated and the
transcription complex for promoter X is sufficient and
that for promoter Y is not sufficient, gene u is ex-
pressed from promoter X, not from promoter Y. For-
mula (2) describes the case that gene u is expressed
from promoter Y. Formula (3) says that if gene u is
not activated, gene u is expressed neither from pro-
moter X nor from promoter Y. Note that we can use
↔ instead of → in the above formulae depending on
our assumptions for a system to be modelled. (The
same argument applies to the other mechanisms.)
The problem is that how we describe the case
when both TC
X
u
and TC
Y
u
are true, i.e. R
+
(u) ∧ TC
X
u
∧
TC
Y
u
→?. The situation is almost the same as the case
when both activators and inhibitors are active for a
gene, which is discussed in our previous work. The
solution depends on the knowledge or assumption we
have on a given network or a problem. We may write
on
X
u
∧ on
Y
u
which means both on
X
u
and on
Y
u
are true,
or write on
X
u
∨ on
Y
u
which means on
X
u
or on
Y
u
are true
(both are true is allowed). If we do not add any clause,
the values of on
X
u
and on
Y
u
are free in this case. An-
other choice is to assume that both TC
X
u
and TC
Y
u
cannot be true simultaneously by adding the clause
G¬(TC
X
u
∧ TC
Y
u
).
For each gene that is regulated by the translated
products of gene u from promoter X/Y, we intro-
duce propositions of the expression levels for u
X
and
u
Y
such as u
X
1
, u
X
2
, . . . , u
Y
1
, u
Y
2
, . . . , and clauses for the
changes of the expression levels of them. These
clauses are the same as those we have for normal
genes.
Readers might wonder what is the difference from
the modelling manner in that we split gene u into the
(virtual) genes u
X
and u
Y
. If we do so, we must dupli-
cate regulating terms R
+
(u) and R
−
(u) into R
+
(u
X
),
R
+
(u
Y
), R
−
(u
X
) and R
−
(u
Y
). For example, if we as-
sume that gene u has two regulators v and w, we need
to introduce the threshold levels of v and w for both
gene u
X
and u
Y
: v
X
u
, v
Y
u
, w
X
u
and w
Y
u
. This blows up
the number of clauses for the changes of the expres-
sion levels of gene u and w. In the above specifica-
tion, however, such blow-ups of the number of propo-
sitions and clauses are avoided.
Mechanism 2. The next mechanism of alternative
splicing is the presence/absence of splicing factors
(SFs). SFs bind to introns or exons and change the
splice sites of an transcribed pre-mRNA of a gene.
SFs can activate or inhibit a certain splice sites, but
the important fact is that splicing is determined by
whether SFs are binding or not. We assume that a
gene u produces two isoforms u
A
(when the SF is
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
154
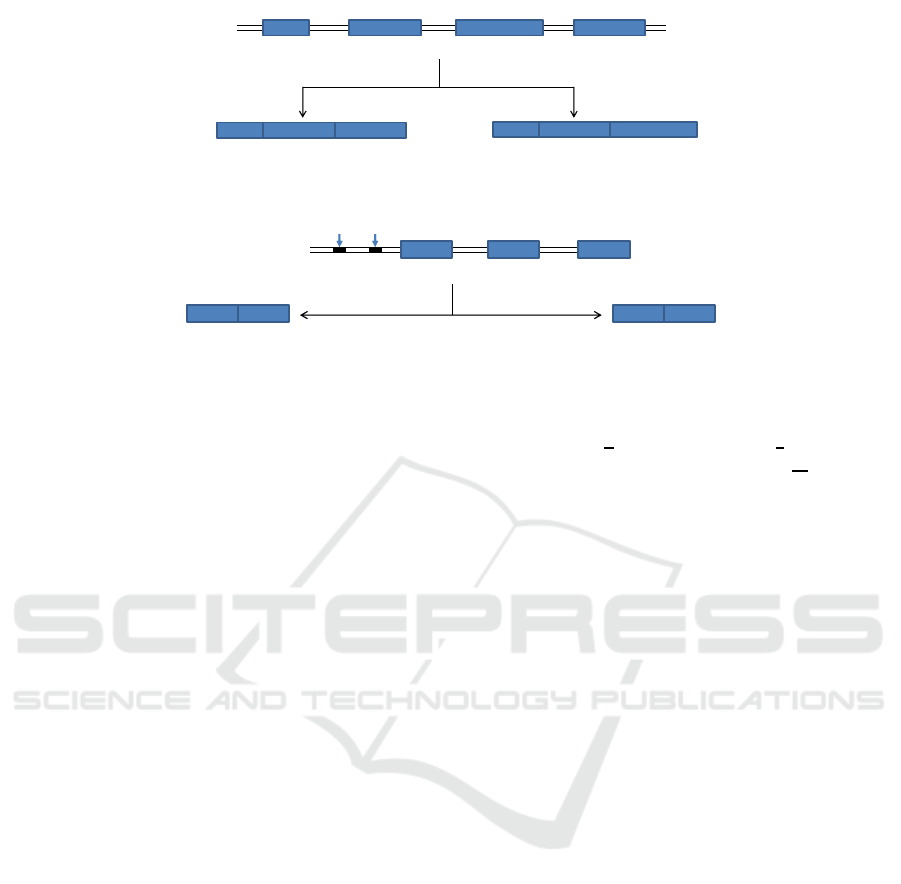
Exon 1 Exon 2 Exon 3 Exon 4
Intron 1 Intron 2 Intron 3
Exon 1 Exon 2 Exon 4 Exon 1 Exon 2 Exon 3
DNA
Figure 4: Alternative splicing produces different mRNAs from a single pre-mRNA.
Exon 1 Exon 2 Exon 3
Exon 1 Exon 3 Exon 2 Exon 3
X
Y
Gene u
From promoter X From promoter Y
Figure 5: Gene u has two promoters X and Y.
binding) and u
B
(when the SF is not binding). We in-
troduce a proposition SF
u
which represents the level
of the SF exceeds the threshold SF
u
upon which the
SF affects on splicing. Then conditions for activat-
ing/inhibiting gene u can be described as:
G(R
+
(u) ∧ SF
u
→ on
A
u
∧ ¬on
B
u
), (4)
G(R
+
(u) ∧ ¬SF
u
→ on
B
u
∧ ¬on
A
u
), (5)
G(R
−
(u) → ¬on
A
u
∧ ¬on
B
u
). (6)
Formula (4) says that if gene u is activated and the
level of the SF is beyond the threshold SF
u
, gene u
produces the isoform u
A
. Formula (5) describes the
case when the level of the SF is not enough to bind
pre-mRNAs of gene u. In this case gene u produces
the isoform u
B
. Formula (6) says that if gene u is not
activated, u does not produce any isoform.
In general, multiple SFs (SF1, SF2, ...) involve
the splicing of a gene u which results in many iso-
forms u
A
, u
B
, u
C
, . . . . By generalising the above for-
mulae we can easily model such complex splicing.
For each combination of effectiveSFs, we specify that
the corresponding isoform is expressed (ON).
4 DEMONSTRATION
In this section we apply our method for modelling al-
ternative splicing described in section 3 to analyse the
network of sex determination in Drosophila (Camara
et al., 2008; Salz and Erickson, 2010).
Genes involved in this sex determination process
are Sxl, tra, tra-2 and dsx. Sxl, tra and dsx have both
male-specific and female-specific splicing. More-
over, Sxl has two promoters – the early promoter
and the late promoter. Sxl is known to have two
female-specific splicing – one from the early pro-
moter and the other from the late promoter. Male-
specific splicing of Sxl occurs only from the late pro-
moter. Thus we have three isoforms from Sxl. We rep-
resent S
e
(from e
arly promoter), S
f
(female-specific
splicing from the late promoter) and S
m
(m
ale-specific
splicing) for each isoform. We similarly write t
f
(female-specific) and t
m
(male-specific) for tra, and
d
f
(female-specific) and d
m
(male-specific) for dsx.
The network controlling sex determination in
Drosophila is illustrated in Fig. 6. First the isoform
S
e
is produced from Sxl by the early promoter. S
e
ac-
tivates female-specific splicing of Sxl itself and pro-
duces the isoform S
f
, which inhibits male-specific
splicing of Sxl and tra. As a result tra produces
female-specific isoform t
f
. This t
f
with tra-2 acti-
vates female-specific splicing of dsx.
To model this network in LTL we introduce the
following propositions.
• on
m
S
, on
f
S
, on
e
S
, on
m
t
, on
f
t
, on
m
d
, on
f
d
: representing
whether the isoforms S
m
, S
f
, S
e
, t
m
, t
f
, d
m
and d
f
are expressed, respectively.
• S
e
S
, S
f
S
, S
f
t
, t
f
d
, t2: these propositions correspond to
whether each isoform is expressed beyond the
threshold level for each activation/inhibition be-
tween genes. S
e
S
corresponds to S
e
+
−→ S
f
, S
f
S
to
S
f
−
−→ S
m
, S
f
t
to S
f
−
−→ t
m
, t
f
d
to t
f
+
−→ d
f
and t2
to tra-2
+
−→ d
f
(see Fig. 6). We consider S
f
S
and
t
f
d
as splicing factors for tra and dsx, respectively.
• TC
E
S
, TC
L
S
: representing whether the levels of tran-
scription complexes of Sxl for the early(E) and
late(L) promoters are sufficient, respectively.
Here we show the essential part, i.e. how the splic-
ing is controlled, of behaviourspecification of the net-
work.
Temporal Logic based Framework to Model and Analyse Gene Networks with Alternative Splicing
155

Sxl
S
m
S
e
S
f
tra
t
m
t
f
dsx
d
m
d
f
tra
-
2
+
-
-
+
+
Figure 6: The network controlling sex determination in Drosophila.
G(TC
E
S
∧ ¬TC
L
S
↔ on
e
S
∧ ¬on
f
S
∧ ¬on
m
S
)∧ (7)
G(S
e
S
∧ TC
L
S
∧ ¬TC
E
S
↔ on
f
S
∧ ¬on
m
S
)∧ (8)
G(¬S
f
S
∧ TC
L
S
∧ ¬TC
E
S
↔ on
m
S
∧ ¬on
f
S
)∧ (9)
G(TC
L
S
↔ (on
m
S
∨ on
f
S
))∧ (10)
G(S
f
S
→ ¬on
m
S
)∧ (11)
G(S
f
t
→ (¬on
m
t
∧ on
f
t
))∧ (12)
G(¬S
f
t
→ (on
m
t
∧ ¬on
f
t
))∧ (13)
G(t2∧t
f
d
→ on
f
d
∧ ¬on
m
d
)∧ (14)
G(t2∧ ¬t
f
d
→ ¬on
f
d
∧ on
m
d
)∧ (15)
G(¬t2 → ¬on
f
d
∧ ¬on
m
d
) ∧ . . . (16)
Formulae (7)-(9) are directly derived from the
mechanism 1 in the previous section. Since we do
not have an explicit regulator for Sxl in the network,
the regulating condition (R
+
(·) in section 3) is empty
in formula (7). Formula (10) reflects the assumption
that isoforms S
m
and S
f
need to be expressed from the
late promoter. Formula (11) stipulates the negative ef-
fect of S
f
to the expression of S
m
. Formulae (12)-(16)
are derived from the mechanism 2 where S
f
t
and t
f
d
as
splicing factors.
For this networkwe check the bistability – female-
specific stability and male-specific stability. The crit-
ical switch to determine this is the female-specific
transcription complex for early promoter of Sxl. If its
intracellular level is sufficient at the initial time, the
cell eventually reaches female-specific stability, oth-
erwise the cell eventually reaches male-specific sta-
bility. This property is described in LTL as:
(TC
E
S
→ FG(on
f
S
∧ on
f
t
∧ on
f
d
))∧
(¬TC
E
S
→ FG(on
m
S
∧ on
m
t
∧ on
m
d
))
in which female(male)-specific stability is written as
that female(male)-specific splicing of the three genes
Sxl, tra and dsx are maintained. Using the LTL satisfi-
ability checker we have the result ’Yes’, which means
that this network surely satisfies the bistability.
We analysed the B¨uchi automaton constructed by
the LTL formula by the LTL satisfiability checker
and investigate the witnesses of the satisfiability
3
.
3
We used GOAL (Tsay et al., 2007) to analyse B¨uchi
Since there are many possible behaviours from all
possible initial state, we extracted a behaviour from
the initial state where gene Sxl is OFF (other genes
are arbitrary) and transcription complex of Sxl for
the early promoter is present/absent. We depict the
behaviours (linear structures) obtained from the au-
tomaton as witnesses in Fig. 7. The states are
represented as vectors of propositional values for
(TC
E
S
, TC
L
S
, on
m
S
, on
f
S
, on
m
t
, on
f
t
, on
m
d
, on
f
d
) in Fig. 7,
where 1 represents true and 0 represents false. For
simplicity the other propositions are omitted, thus the
edge in the figure does not necessarily corresponds
the one atomic step of the original behaviour because
the states in which only the values of the omitted
propositions differ are identified.
In both Fig. 7(a) and (b), after some initial pertur-
bation, the network reaches the stable state in which
all the sex-specific splicing is maintained. Note that
this is just instances of the possible behaviours hap-
pened to be produced by the automaton as witnesses.
The behaviours include some interesting features: the
transcription for the late promoter of Sxl is some-
times cut off in the female-specific behaviour (a) and
the transcription factor for the early promoter of Sxl,
which is known to be a female-specific transcription
complex, is once produced in the male-specific be-
haviour (b), nevertheless, the network reaches the
final desirable states (only sex-specific splicing are
maintained). This can be interpreted that the network
has homeostasis against the perturbation on the tran-
scription complexes for Sxl.
Compared to Fig. 7 (b), the interpretation of
(a) might be a bit difficult: especially the step from
the state 00001000 to the last state 01010101 looks
somewhat mysterious. The key to understand this
behaviour is the previous two states (1111
1000 and
01011000) where gene Sxl is expressed in the female-
specific isoform (underlined). Therefore the isoform
is sufficiently stored while the supply of the transcrip-
tion complex for the late promoter is instantaneously
suspended (at the state 00001000). Thus the female-
specific splicing of Sxl is re-started once the supply
of the transcription complex for the late promoter is
resumed and maintained (at the last state 01010101).
automata.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
156
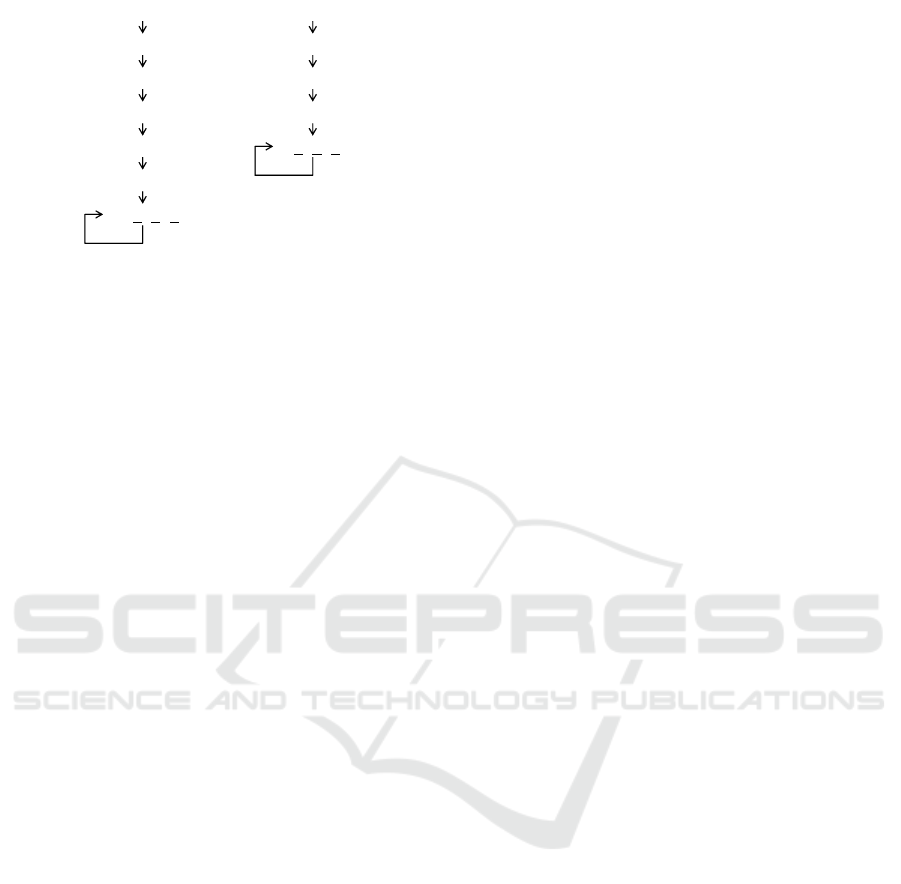
10000101
01010100
00000101
01101011
(a) (b)
11111000
01101010
11111010
01011000
00001000
00000100
01010101
01101010
Figure 7: Example behaviours of the sex determina-
tion network in Drosophila. For simplicity the vec-
tor only consists of the values of the propositions
(TC
E
S
, TC
L
S
, on
m
S
, on
f
S
, on
m
t
, on
f
t
, on
m
d
, on
f
d
). (a) A female-
specific behaviour. Underlined are female-specific iso-
forms. (b) A male-specific behaviour. Underlined are male-
specific isoforms.
Next we check another property: this network is
not able to keep female- and male-specific splicing
simultaneously. This property is formally written as:
FG(on
f
S
∧ on
f
t
∧ on
f
d
∧ on
m
S
∧ on
m
t
∧ on
m
d
)
The result is ’No’, as we expected. There is no
such behaviour in the possible behaviours of the net-
work controlling sex determination. Both verifica-
tions show that this network ensures the sex determi-
nation.
The sex determination of Drosophila is also mod-
elled and analysed in (Louis et al., 2003). In that
work, the authors model the behaviour of the expres-
sion of gene Sxl and analyse how the bistable female-
specific and male-specific differences arise. They use
ODE models for the transcription of Sxl from theearly
promoter and probabilistic models for that of the late
promoter. The two models are combined to employ
the overall analysis. Their model is elaborated and re-
quires deep knowledge in molecular mechanisms. In
addition, the mathematical inference of unavailable
kinetic parameters is required. For simplicity they
abstracted the downstream genes of Sxl such as tra
and dsx. We guess the reason of this simplification
in the modelling and analysis as that the cascading
the quantitative model makes the entire model very
sensitive to the changes of parameter values in each
stage and to ‘correct’ inference of the uncertain pa-
rameters is more crucial to reproduce the sex determi-
nation. In contrast, our qualitative framework allows
us to model the network concisely with the same con-
clusion (bistability of the network). The downstream
genes of Sxl are also included in the analysis without
difficulty. We must note, however, that their quanti-
tative model enables the robustness analysis such that
how the sex determination works against the modula-
tion of gene doses. Due to qualitativeness of our for-
malism, such analysis is infeasible in our framework.
We, however, showed that both female-specific and
male-specific gene expressions cannot be maintained,
i.e. the possible behaviours of the network does not
contain such behaviours. To prove this with quanti-
tative model is very difficult since we need to test all
combinations of possible quantitative parameters and
confirm that such behaviours are never produced.
(Fear et al., 2015) modelled the sex determina-
tion regulatory network of Drosophila using struc-
tural equation models. The purpose of their work is
to infer statistically likely links between genes in the
known network or to find new genes which can likely
be included in the network, rather than to investigate
how the sex determination is ensured by the network.
Although the aim of modelling the network is differ-
ent from ours, the prediction of plausible extension of
the known network is interesting aspect of systems bi-
ology. This line of research will be future work in our
formalism. Whereas their method for finding plausi-
ble extension is brute force: they enumerate all possi-
ble interactions and insertion of new genes in all pos-
sible locations in the graph, in our framework some
logical inference may be helpful to find plausible ex-
tensions of the network instead of using brute force.
5 CONCLUSION
In this paper we presented a qualitative framework to
model and analyse gene networks using linear tem-
poral logic. We studied molecular mechanisms of al-
ternative splicing and showed how such mechanisms
are modelled in our framework. As a demonstra-
tion, we modelled the network of sex determination
in Drosophila and checked the sex-specific bistability
of the network.
Since this work is still at a theoretical stage, we
are investigating applications of our frameworkto real
biological problems. For this we are to develop (semi-
)automatic modelling method from gene regulation
information. Compared to quantitative approaches
which need manual parameter inference or tuning, a
(semi-)automatic model construction of a gene regu-
latory network ismore feasible in ourframework. The
start point will be to devise a machine readable uni-
form presentation of splicing networks. One promis-
ing approach is to extend SBML Qualitative Models
Package (Chaouiya et al., 2013).
Temporal Logic based Framework to Model and Analyse Gene Networks with Alternative Splicing
157

ACKNOWLEDGEMENT
This work was supported by JSPS KAKENHI Grant
Number 26730153.
REFERENCES
Aoshima, T. (2003). On a verification System for Reactive
System Specifications. PhD thesis, Tokyo Institute of
Technology.
Batt, G., Salah, R. B., and Maler, O. (2007). On timed
models of gene networks. In FORMATS 2007, volume
4763 of LNCS, pages 38–52.
Camara, N., Whitworth, C., and Van Doren, M. (2008).
The creation of sexual dimorphism in the Drosophila
soma. Curr. Top. Dev. Biol., 83:65–107.
Chaouiya, C., B´erenguier, D., Keating, S. M., Naldi, A.,
van Iersel, M. P., Rodriguez, N., Dr¨ager, A., B¨uchel,
F., Cokelaer, T., Kowal, B., Wicks, B., Gonc¸alves, E.
J. V., Dorier, J., Page, M., Monteiro, P. T., von Kamp,
A., Xenarios, I., de Jong, H., Hucka, M., Klamt, S.,
Thieffry, D., Nov`ere, N. L., Saez-Rodriguez, J., and
Helikar, T. (2013). SBML qualitative models: a model
representation format and infrastructure to foster in-
teractions between qualitative modelling formalisms
and tools. BMC Systems Biology, 7:135.
Ciocchetta, F. and Hillston, J. (2009). Bio-PEPA: A frame-
work for the modelling and analysis of biological sys-
tems. Theor. Comput. Sci., 410:3065–3084.
David, C. and Manley, J. (2008). The search for alternative
splicing regulators: new approaches offer a path to a
splicing code. Genes Dev., 22(3):279–285.
Fear, J., Arbeitman, M. N., Salomon, M., Dalton, J., Tower,
J., Nuzhdin, S. V., and McIntyre, L. M. (2015). The
wright stuff: reimagining path analysis reveals novel
components of the sex determination hierarchy in
drosophila melanogaster. BMC Systems Biology, 9:53.
Heiner, M., Gilbert, D. R., and Donaldson, R. (2008). Petri
nets for systems and synthetic biology. In SFM 2008,
volume 5016 of LNCS, pages 215–264.
Hertel, K. (2008). Combinatorial control of exon recogni-
tion. J. Biol. Chem., 283(3):1211–1215.
Ito, S., Hagihara, S., and Yonezaki, N. (2014). A qualita-
tive framework for analysing homeostasis in gene net-
works. In Proceedings of BIOINFORMATICS 2014,
pages 5–16.
Ito, S., Ichinose, T., Shimakawa, M., Izumi, N., Hagihara,
S., and Yonezaki, N. (2013a). Modular analysis of
gene networks by linear temporal logic. J. Integrative
Bioinformatics, 10(2).
Ito, S., Ichinose, T., Shimakawa, M., Izumi, N., Hagihara,
S., and Yonezaki, N. (2013b). Qualitative analysis of
gene regulatory networks using network motifs. In
Proceedings of BIOINFORMATICS 2013, pages 15–
24.
Ito, S., Ichinose, T., Shimakawa, M., Izumi, N., Hagihara,
S., and Yonezaki, N. (2015). Qualitative analysis of
gene regulatory networks by temporal logic. Theor.
Comput. Sci., 594(23):151–179.
Ito, S., Izumi, N., Hagihara, S., and Yonezaki, N. (2010).
Qualitative analysis of gene regulatory networks by
satisfiability checking of linear temporal logic. In Pro-
ceedings of BIBE 2010, pages 232–237.
Kornblihtt, A. (2005). Promoter usage and alternative splic-
ing. Curr. Opin. Cell Biol., 17(3):262–268.
Louis, M., Holm, L., S´anchez, L., and Kaufman, M. (2003).
A theoretical model for the regulation of sex-lethal,
a gene that controls sex determination and dosage
compensation in drosophila melanogaster. Genetics,
165(3):1355–84.
Matlin, A., Clark, F., and Smith, C. (2005). Understanding
alternative splicing: towards a cellular code. Nat. Rev.
Mol. Cell Biol., 6(5):386–398.
Palsson, B. (2000). The challenges of in silico biology. Nat.
Biotechnol., 18:1147–50.
Salz, H. K. and Erickson, J. W. (2010). Sex determination
in Drosophila: The view from the top. Fly, 4:60–70.
Thomas, R. (1991). Regulatory networks seen as asyn-
chronous automata: A logical description. J. Theor.
Biol., 153(1):1–23.
Tsay, Y.-K., Chen, Y.-F., Tsai, M.-H., Wu, K.-N., and Chan,
W.-C. (2007). GOAL: a graphical tool for manipu-
lating B¨uchi automata and temporal formulae. In Pro-
ceedings of the 13th international conference on Tools
and algorithms for the construction and analysis of
systems, volume 4424 of TACAS’07, pages 466–471,
Berlin, Heidelberg. Springer-Verlag.
Vardi, M. Y. and Wolper, P. (1994). Reasoning about infinite
computations. Inf. Comput., 115:1–37.
Wen, J. (2013). Computational modeling and inference of
alternative splicing regulation. PhD thesis, University
of Miami.
BIOINFORMATICS 2016 - 7th International Conference on Bioinformatics Models, Methods and Algorithms
158
